Prerequisites
- Access to an RCAC cluster (Negishi or Gautschi)
- Familiarity with the Linux command line (cd, ls, mkdir)
- A terminal session via Open OnDemand or SSH
Prerequisites
Objectives
Quality control is the most important step you can take before starting any genomics analysis. The principle is simple: garbage in, garbage out. If your raw sequencing data has problems such as adapter contamination, low-quality bases, or reads from the wrong organism, every downstream result is compromised. The worst part is that these problems often propagate silently. Your pipeline will still produce output files. You will still get variant calls, expression counts, or an assembly. They will just be wrong.
QC is not a one-time checkbox at the beginning of your workflow. You should check quality at every stage:
The cost of skipping QC is real: wasted compute hours processing bad data, false positive results that don’t replicate, and in the worst case, retracted papers. The good news is that QC is fast and easy. Fifteen minutes of checking quality can save you weeks of troubleshooting.
Recommended starting point. Open OnDemand provides a web portal with a file browser, terminal, job submission forms, and interactive apps like JupyterLab and RStudio. No software to install.
Navigate to the OOD portal
Go to https://gateway.<cluster>.rcac.purdue.edu and log in with your Purdue (BoilerKey) credentials.
| Cluster | OOD URL |
|---|---|
| Gautschi | gateway.gautschi.rcac.purdue.edu |
| Negishi | gateway.negishi.rcac.purdue.edu |
| Bell | gateway.bell.rcac.purdue.edu |
| Gilbreth | gateway.gilbreth.rcac.purdue.edu |
Tour the dashboard
After login you will see the OOD dashboard with these key sections:
Open a terminal
Click Clusters in the top menu and select the cluster shell access (e.g., Gautschi Shell Access). A terminal opens in your browser. You are now on a login node.
SSH gives you direct terminal access. It is built into macOS and Linux. On Windows, use PowerShell, WSL, PuTTY, or MobaXterm.
ssh <boilerid>@gautschi.rcac.purdue.eduReplace <boilerid> with your Purdue career account username. For other clusters, replace gautschi with the cluster name.
ThinLinc provides a full Linux desktop environment, ideal for GUI-based tools like IGV, CellProfiler, MEGA, or any application that needs a graphical interface.
Install the ThinLinc client
Download and install the ThinLinc client for your operating system from the ThinLinc download page.
Connect to the cluster
Open the ThinLinc client and enter the connection details:
| Field | Value |
|---|---|
| Server | desktop.<cluster>.rcac.purdue.edu |
| Username | Your Purdue career account (BoilerID) |
| Password | Your Purdue password |
For example, to connect to Gautschi: desktop.gautschi.rcac.purdue.edu
Authenticate
After entering your credentials, you will be prompted for Duo two-factor authentication. Approve the push notification or enter the code.
Use the desktop
You now have a full Linux desktop. Open a terminal from the Applications menu to run commands, or launch GUI applications directly.
You can also access ThinLinc through a web browser (no client install needed) at https://desktop.<cluster>.rcac.purdue.edu/. The native client generally provides a smoother experience.
For detailed ThinLinc documentation, see the RCAC ThinLinc guide.
cd $RCAC_SCRATCHmkdir -p genomics_exchange/session6cd genomics_exchange/session6RCAC provides bioinformatics software through the module system. Always start with a clean environment:
module --force purgeWe will load specific tool modules as needed in each section below.
FastQC is the standard tool for assessing the quality of sequencing data. It takes FASTQ files as input and produces an HTML report with visualizations of key quality metrics.
Before running FastQC, it helps to understand what a FASTQ file looks like. Each read in a FASTQ file consists of four lines:
@read_name <- Read identifierACGTACGTACGT... <- DNA sequence+ <- SeparatorIIIIIHHHGG... <- Quality scores (one character per base)The quality scores on line 4 are encoded as ASCII characters. Each character represents a Phred quality score: a measure of how confident the sequencer is that it read each base correctly.
| Phred Score | Error Probability | Accuracy |
|---|---|---|
| Q10 | 1 in 10 | 90% |
| Q20 | 1 in 100 | 99% |
| Q30 | 1 in 1,000 | 99.9% |
| Q40 | 1 in 10,000 | 99.99% |
Modern Illumina sequencers typically produce data with Q30 or higher for most bases. Q20 is generally considered the minimum acceptable quality.
In paired-end sequencing, both ends of each DNA fragment are read, producing two files per sample: R1 (forward read) and R2 (reverse read). This gives you more information about where each fragment came from in the genome and is the most common approach for Illumina sequencing.
Load the FastQC module:
module load biocontainers fastqc/0.12.1Create an output directory and run FastQC:
mkdir -p fastqc_rawfastqc -t 4 -o fastqc_raw/ raw_reads/sample1_R1.fastq.gz raw_reads/sample1_R2.fastq.gz-t 4: use 4 threads (one per file, speeds things up)-o fastqc_raw/: write output to this directoryFor multiple samples, loop over all files:
for fq in raw_reads/*.fastq.gz; do fastqc -t 4 -o fastqc_raw/ "$fq"doneView the HTML report. You can open it through the Open OnDemand file browser (navigate to the file and click on it) or download it to your laptop:
scp boilerid@negishi.rcac.purdue.edu:$RCAC_SCRATCH/genomics_exchange/session6/fastqc_raw/sample1_R1_fastqc.html .FastQC evaluates 12 quality modules. Each module gets a pass (green check), warn (orange triangle), or fail (red X) status. The table below explains what each module measures and how to interpret it.
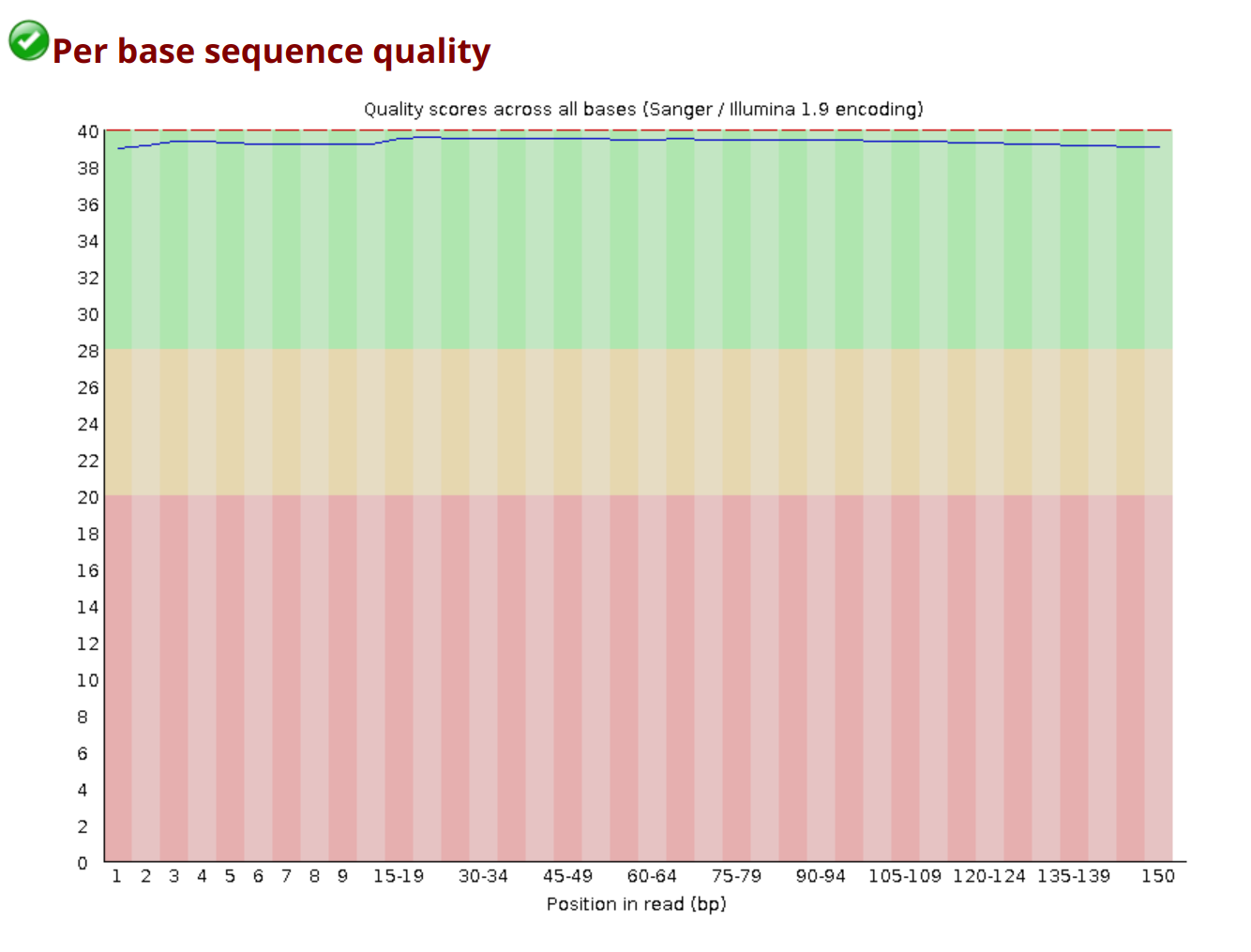
A healthy per-base quality plot looks like this: scores stay in the green zone (Q28+) across the entire read, with only a small dip at the 3’ end.
| Module | What It Measures | Good | Bad | Common False Flags |
|---|---|---|---|---|
| Basic Statistics | Read count, length, GC% | Consistent with expectations | Unexpected read count or length | Rarely triggers false flags |
| Per Base Sequence Quality | Phred scores at each position along the read | Q28+ across most positions; slight drop at 3’ end is normal | Q < 20 across large portions of the read | Rarely false; always investigate |
| Per Tile Sequence Quality | Quality variation across flow cell tiles | Uniform blue heatmap | Hot spots (yellow/red patches) indicate flow cell problems | Rare |
| Per Sequence Quality Scores | Distribution of average quality per read | Single peak at Q30-Q37 | Bimodal distribution (two peaks) | Rare |
| Per Base Sequence Content | %A, %T, %G, %C at each position | Roughly equal A/T and G/C; flat lines | Strong bias throughout the read | First 10-15 bases often wobble due to random hexamer priming. This is normal |
| Per Sequence GC Content | Distribution of GC% across all reads | Smooth bell curve matching your organism | Bimodal curve = contamination from another organism | Rarely false; investigate if unexpected |
| Per Base N Content | Percentage of N (unknown) bases at each position | Near 0% everywhere | Spikes of N calls indicate sequencer problems | Rare |
| Sequence Length Distribution | Distribution of read lengths | All reads same length (pre-trimming) | Unexpected variation | After trimming, variation is expected and normal |
| Sequence Duplication Levels | How many reads appear more than once | Most reads unique (low duplication) | High duplication in WGS = low library complexity | RNA-seq: high duplication is normal (highly expressed genes). Amplicon: high duplication is expected. |
| Overrepresented Sequences | Sequences making up > 0.1% of reads | None or very few | Adapter sequences, rRNA contamination | RNA-seq: abundant transcripts trigger this. Not always a problem. |
| Adapter Content | Cumulative adapter contamination at each position | Near 0% throughout | Rising curves at read ends = adapter read-through | Rarely false; needs trimming if present |
| Kmer Content | Overrepresented short sequences at specific positions | No enrichment | Enrichment at specific positions = primer or adapter contamination | First few bases often enriched due to priming bias, usually normal |
The cartoons below (by Zandra Selina, CC BY 4.0) show what each module looks like for healthy data and for the most common failure modes. Use them as a quick mental lookup when you open a real report.
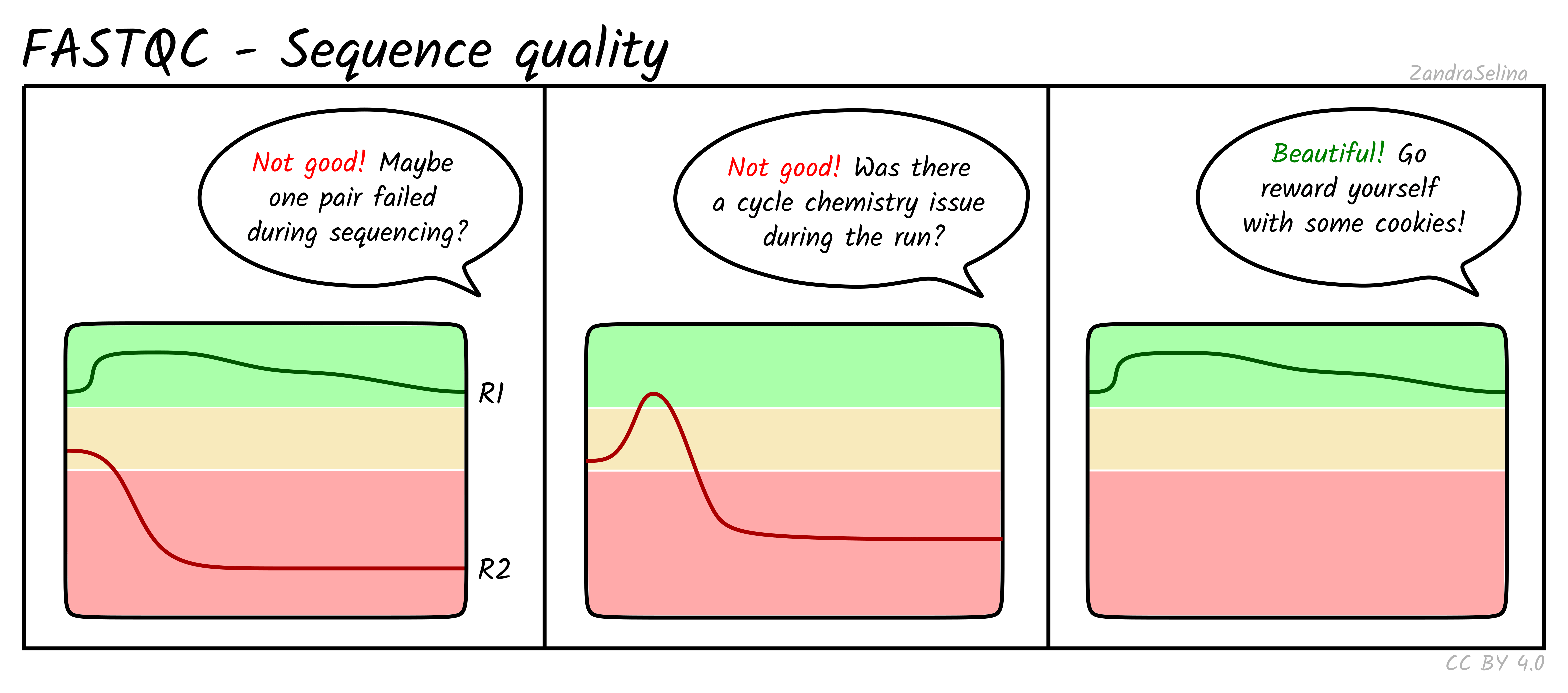
R2 is expected to be slightly worse than R1, but a sharp collapse or a mid-read dip points to a sequencing run problem rather than a library issue.
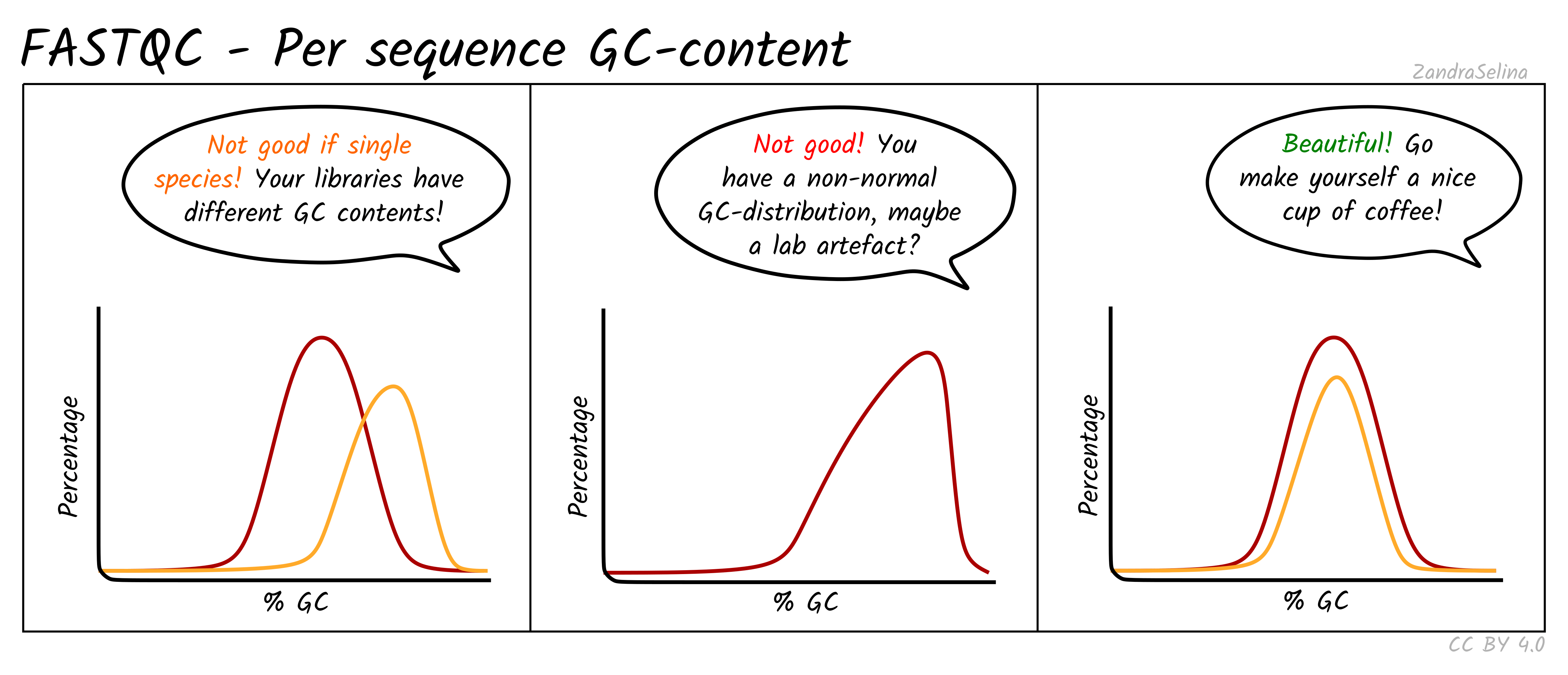
For RNA-seq, mild deviations from a bell curve are normal because expressed transcripts are not GC-neutral. A clearly bimodal curve, however, suggests cross-species contamination.
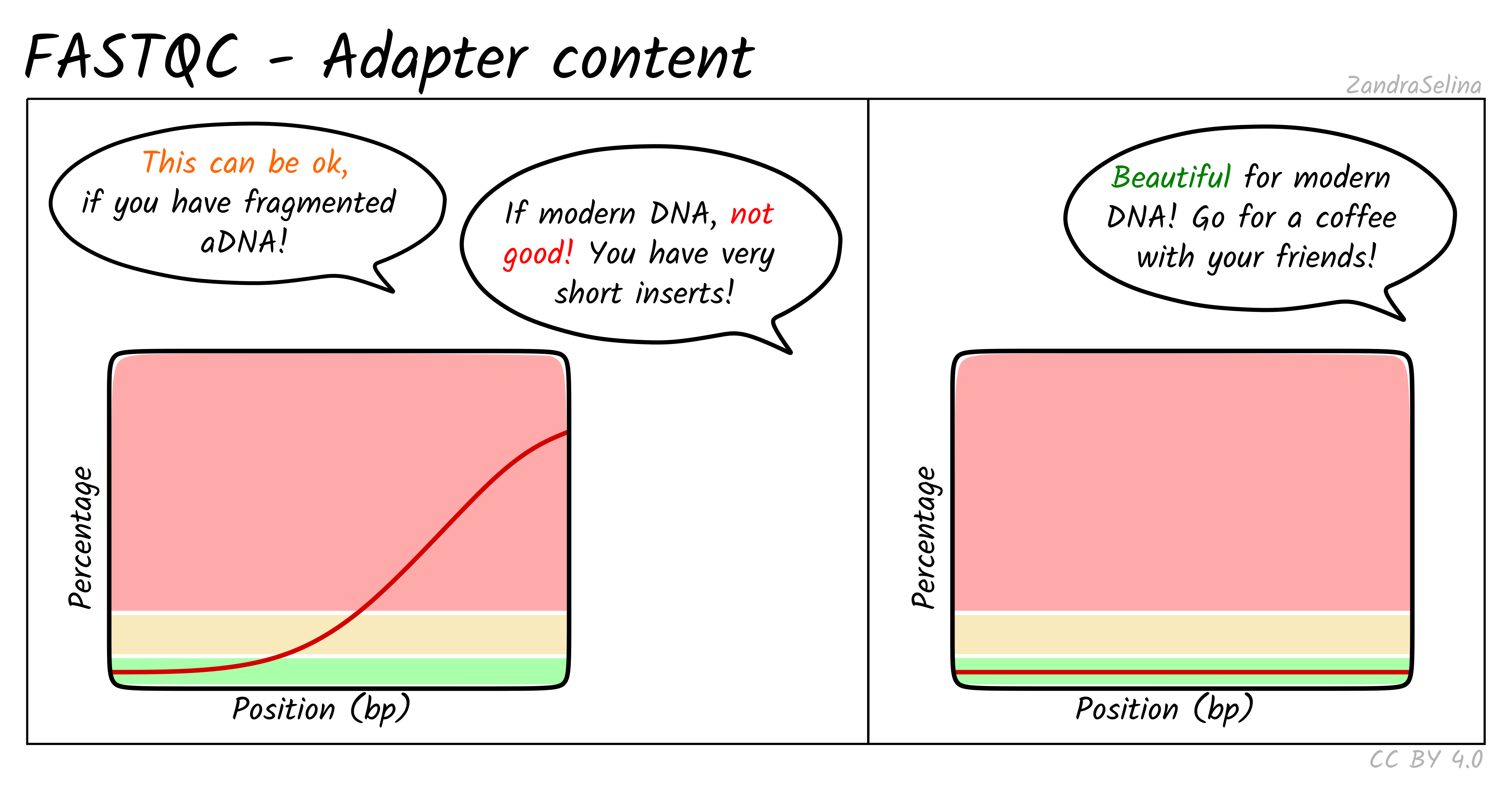
A rising curve at read ends means inserts are shorter than the read length. This is expected for ancient or degraded DNA, but for modern libraries it indicates short fragments that may benefit from trimming.
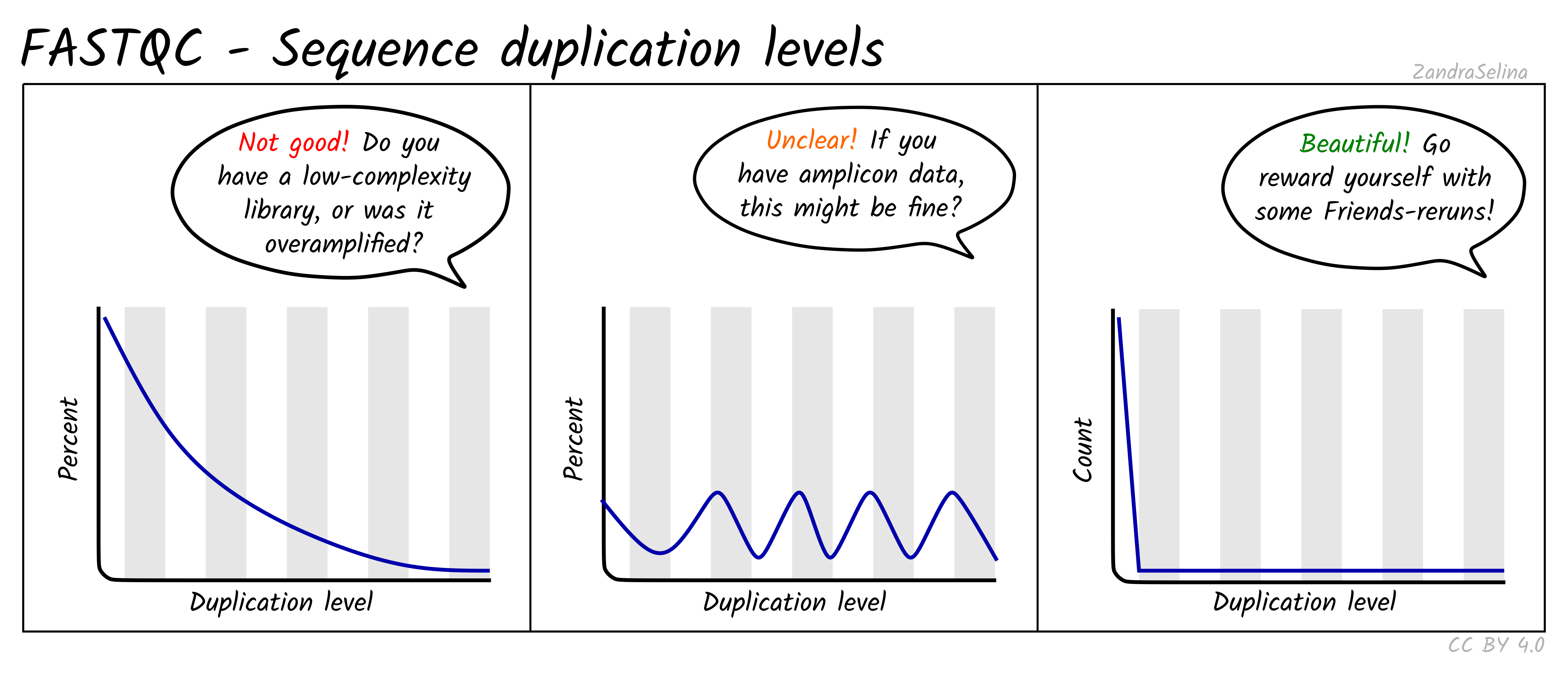
High duplication is expected for RNA-seq (highly expressed genes) and amplicon data, but a heavy tail in WGS points to overamplification or low input material.
When you have many samples, opening individual FastQC reports one by one is tedious. MultiQC aggregates results from FastQC (and many other tools) into a single interactive HTML report.
Load MultiQC:
module load biocontainers multiqc/1.25.1Run MultiQC on all FastQC results:
mkdir -p multiqc_rawmultiqc fastqc_raw/ -o multiqc_raw/Open multiqc_raw/multiqc_report.html in your browser.
The power of MultiQC is in spotting outliers. When all samples are plotted together:
The MultiQC status check heatmap gives you a one-glance view of which modules pass, warn, or fail across all samples:
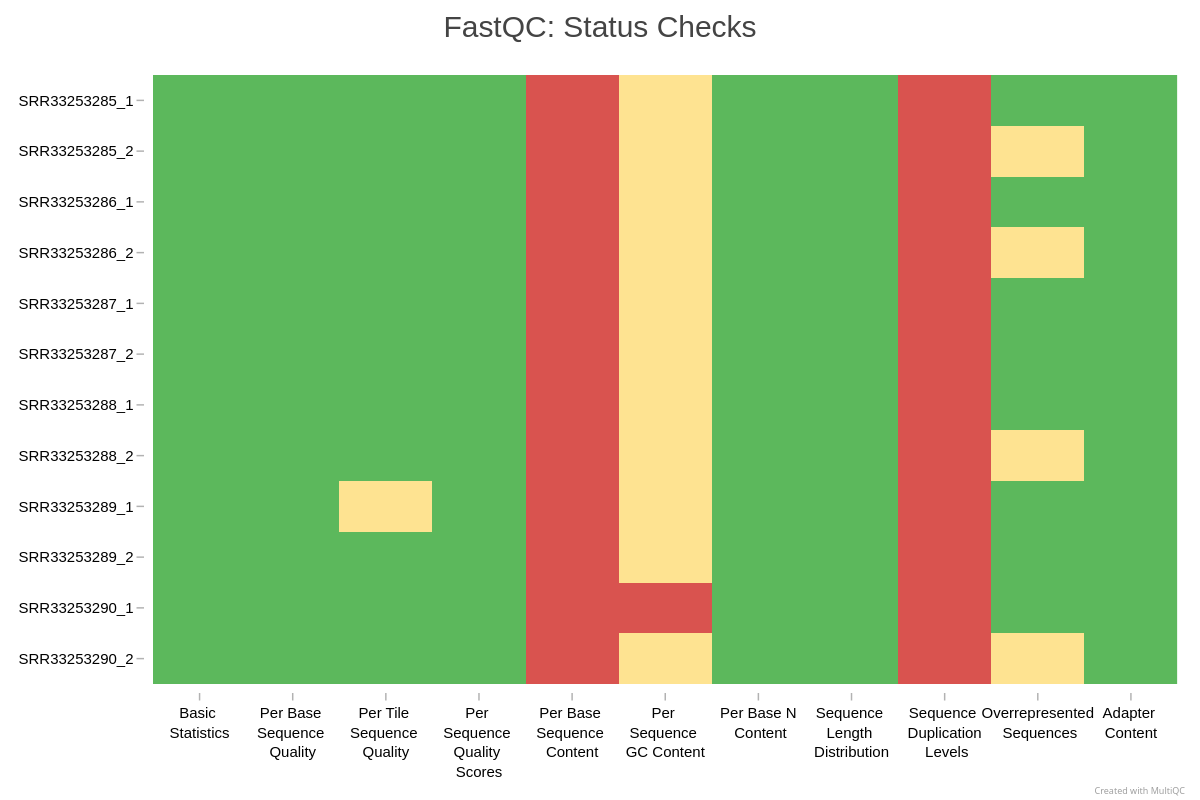
In this example, every sample fails Per Base Sequence Content and Sequence Duplication Levels. For RNA-seq this is expected: random hexamer priming biases the first ~12 bases, and highly expressed transcripts inflate duplication. The Per Sequence GC Content warnings reflect transcriptome composition rather than a library defect. None of these are reasons to discard data.
fastp is an all-in-one tool for quality filtering, adapter trimming, and read preprocessing. It is fast, handles paired-end data well, and produces its own HTML report.
Adapters are short synthetic DNA sequences added during library preparation. They allow the DNA fragments to attach to the sequencer’s flow cell. Adapters are not part of your organism’s genome. If a DNA fragment is shorter than the read length, the sequencer reads past the insert and into the adapter on the other side. These adapter sequences need to be removed before analysis.
Load fastp:
module load biocontainers fastp/0.24.0Run fastp on a paired-end sample:
mkdir -p trimmed fastp_reportsfastp \ -i raw_reads/sample1_R1.fastq.gz \ -I raw_reads/sample1_R2.fastq.gz \ -o trimmed/sample1_R1.trimmed.fq.gz \ -O trimmed/sample1_R2.trimmed.fq.gz \ --qualified_quality_phred 20 \ --length_required 50 \ --detect_adapter_for_pe \ --thread 4 \ --html fastp_reports/sample1.html \ --json fastp_reports/sample1.jsonOpen fastp_reports/sample1.html in your browser to view the before/after quality report.
| Parameter | Default | Description | When to Change |
|---|---|---|---|
--qualified_quality_phred | 15 | Bases below this quality are “unqualified” | Use 20 for standard analysis; 30 for variant calling where accuracy is critical |
--unqualified_percent_limit | 40 | Drop read if this % of bases are unqualified | Lower for stricter filtering |
--length_required | 15 | Discard reads shorter than this after trimming | Use 50 for most Illumina data; adjust based on your aligner’s minimum |
--detect_adapter_for_pe | off | Auto-detect adapters via paired-end overlap | Always enable for paired-end data |
--adapter_sequence | auto | Specify adapter sequence manually | Only if auto-detection fails |
--cut_front | off | Sliding window trimming from 5’ end | Enable for data with 5’ quality issues |
--cut_tail | off | Sliding window trimming from 3’ end | Enable for data with 3’ quality dropoff |
--cut_window_size | 4 | Window size for sliding window trimming | Larger window = more aggressive trimming |
--cut_mean_quality | 20 | Quality threshold for sliding window | Match to your --qualified_quality_phred |
--trim_poly_g | auto | Trim poly-G tails | Important for NovaSeq/NextSeq (two-color chemistry artifacts) |
--correction | off | Overlap-based error correction for PE reads | Enable for variant calling workflows |
--thread | 1 | Number of processing threads | Use 4 for good performance; fastp is fast even single-threaded |
The fastp report shows:
After trimming, run FastQC again on the trimmed reads and use MultiQC to compare raw vs. trimmed results side by side.
Run FastQC on trimmed reads:
mkdir -p fastqc_trimmedfor fq in trimmed/*.trimmed.fq.gz; do fastqc -t 4 -o fastqc_trimmed/ "$fq"doneRun MultiQC on everything (raw FastQC + trimmed FastQC + fastp reports):
mkdir -p multiqc_combinedmultiqc fastqc_raw/ fastqc_trimmed/ fastp_reports/ -o multiqc_combined/Open multiqc_combined/multiqc_report.html and compare:
There is no single universal threshold. What “good enough” means depends on your downstream analysis, your organism, and your experimental design. Here are general guidelines:
| Metric | Good | Acceptable | Investigate |
|---|---|---|---|
| % bases Q30+ (after trim) | > 90% | 80-90% | < 80% |
| % adapter content (after trim) | < 1% | 1-5% | > 5% |
| % reads retained after trimming | > 95% | 85-95% | < 85% |
| Duplication rate (WGS) | < 15% | 15-30% | > 30% |
| GC content distribution | Unimodal, matches organism | Slight shoulder | Bimodal |
Use this table to decide what action to take based on your QC results:
| Scenario | Action |
|---|---|
| Adapter content > 5% | Trim with fastp (--detect_adapter_for_pe). Re-run FastQC to confirm removal. |
| Mean quality < Q25 | Trim with fastp quality filtering. If still poor, consider re-sequencing. |
| Quality drops sharply at 3’ end | Normal for Illumina. Light trimming with fastp is sufficient. |
| GC content bimodal | Investigate contamination. Check for mixed species. Consider decontamination tools (e.g., Kraken2). |
| High duplication in WGS | Low library complexity. May need re-sequencing with more input DNA. |
| High duplication in RNA-seq | Normal: highly expressed genes produce duplicate reads. Not a QC failure. |
| Overrepresented adapter sequences | Trim with fastp. Confirm removal with FastQC. |
| Overrepresented biological sequences | Check what the sequence is (BLAST it). May be rRNA contamination in RNA-seq. |
| Per base sequence content wobbles at 5’ end | Normal: random hexamer priming bias. No action needed. |
| Very low read count for one sample | Check library prep. May need re-sequencing. Exclude from analysis if insufficient depth. |
Quality control does not end with read trimming. Each stage of your analysis has its own QC metrics.
After aligning reads to a reference genome (with BWA-MEM2, STAR, HISAT2, etc.), check:
module load biocontainers samtools/1.21| Tool / Command | What It Shows |
|---|---|
samtools flagstat sample.bam | Total reads, mapped reads, properly paired reads, duplicates |
samtools stats sample.bam | Comprehensive alignment statistics including insert size, error rates |
picard CollectAlignmentSummaryMetrics | Mapping rate, mismatch rate, by read group |
picard CollectInsertSizeMetrics | Insert size distribution (should match library prep) |
Key metrics:
| Metric | Expected Value | What It Means |
|---|---|---|
| Ti/Tv ratio (WGS, human) | 2.0-2.1 | Lower values suggest false positive SNP calls |
| Ti/Tv ratio (exome, human) | 2.8-3.0 | Exonic regions have higher Ti/Tv |
| Het/Hom ratio (human) | 1.5-2.0 | Very high = contamination; very low = inbreeding or sample issue |
After alignment with STAR or HISAT2:
All commands used in this session in one place:
# --- Setup ---module --force purgecd $RCAC_SCRATCHmkdir -p genomics_exchange/session6cd genomics_exchange/session6
# --- FastQC on raw reads ---module load biocontainers fastqc/0.12.1mkdir -p fastqc_rawfastqc -t 4 -o fastqc_raw/ raw_reads/*.fastq.gz
# --- MultiQC on raw FastQC results ---module load biocontainers multiqc/1.25.1mkdir -p multiqc_rawmultiqc fastqc_raw/ -o multiqc_raw/
# --- Trim with fastp ---module load biocontainers fastp/0.24.0mkdir -p trimmed fastp_reportsfastp \ -i raw_reads/sample1_R1.fastq.gz \ -I raw_reads/sample1_R2.fastq.gz \ -o trimmed/sample1_R1.trimmed.fq.gz \ -O trimmed/sample1_R2.trimmed.fq.gz \ --qualified_quality_phred 20 \ --length_required 50 \ --detect_adapter_for_pe \ --thread 4 \ --html fastp_reports/sample1.html \ --json fastp_reports/sample1.json
# --- FastQC on trimmed reads ---mkdir -p fastqc_trimmedfastqc -t 4 -o fastqc_trimmed/ trimmed/*.trimmed.fq.gz
# --- Combined MultiQC report ---mkdir -p multiqc_combinedmultiqc fastqc_raw/ fastqc_trimmed/ fastp_reports/ -o multiqc_combined/
# --- Alignment QC (after aligning with BWA-MEM2 or STAR) ---module load biocontainers samtools/1.21samtools flagstat aligned/sample1.sorted.bamsamtools stats aligned/sample1.sorted.bam > sample1.stats.txtUp next: Session 7 (April 21, 2026): Reproducible bioinformatics using Nextflow. We will learn how to wrap QC, alignment, and analysis into reproducible pipelines using Nextflow and nf-core.