Assemble Mitochondrial Genomes from Long Reads
A short guide to assemble mitochondrial genomes from long reads (PacBio HiFi) using MitoHiFi program
1. Installation
Section titled “1. Installation”Using the docker image, we can run MitoHiFi on any system that supports docker. Singularity can also be used to run the docker image on HPC systems. All RCAC systems support Singularity.
singularity pull docker://ghcr.io/marcelauliano/mitohifi:masterThis will create mitohifi-master.sif file in the current directory.
2. Running MitoHiFi
Section titled “2. Running MitoHiFi”To run MitoHiFi, we need HiFi reads. We will download sample datasets from PacBio website. For this specific tutorial, we will use the PacBio HiFi data for maize B73 genome.
wget https://downloads.pacbcloud.com/public/revio/2023Q1/maize-B73-rep1/m84006_221229_002525_s1.hifi_reads.bamFirst step is to convert the HiFi reads in bam format to fasta format. We can use samtools for this.
ml biocontainersml samtoolssamtools fasta \ -threads ${SLURM_CPUS_ON_NODE} \ m84006_221229_002525_s1.hifi_reads.bam > maize_B73_hifi.fastaWe will also need a reference mitochondrial genome. MitoHifi provides a script to download the reference mitochondrial genome for maize.
singularity exec mitohifi_master.sif findMitoReference.py \ --species "Zea mays subsp. mays" \ --email ${USER}@purdue.edu \ --outfolder ./maize/ \ --type mitochondrion \ --min_length 14000This command will download NC_007982.1.fasta and NC_007982.1.gb in the maize directory that you will use for -f and -g options in the next step.
Next, we can run MitoHiFi on the fasta file
singularity exec mitohifi_master.sif mitohifi.py \ -r maize-hifi.fasta \ -f maize/NC_007982.1.fasta \ -g maize/NC_007982.1.gb \ -t ${SLURM_CPUS_ON_NODE} \ -a plant \ -p 80 \ -o 1The options to consider are:
-t: Number of threads, using${SLURM_CPUS_ON_NODE}to get the number of CPUs on the node you requested-a: organism group, whetheranimal,plant, orfungi-p: Percentage of query in the blast match with close related mito, default is 50-o: Genetic-code- 1: The Standard Code
- 2: The Vertebrate Mitochondrial Code
- 3: The Yeast Mitochondrial Code
- 4: The Mold, Protozoan, and Coelenterate Mitochondrial Code and the Mycoplasma/Spiroplasma Code
- 5: The Invertebrate Mitochondrial Code
- 6: The Ciliate, Dasycladacean and Hexamita Nuclear Code
- 9: The Echinoderm and Flatworm Mitochondrial Code
- 10: The Euplotid Nuclear Code
- 11: The Bacterial, Archaeal and Plant Plastid Code
- 12: The Alternative Yeast Nuclear Code
- 13: The Ascidian Mitochondrial Code
- 14: The Alternative Flatworm Mitochondrial Code
- 16: Chlorophycean Mitochondrial Code
- 21: Trematode Mitochondrial Code
- 22: Scenedesmus obliquus Mitochondrial Code
- 23: Thraustochytrium Mitochondrial Code
- 24: Pterobranchia Mitochondrial Code
- 25: Candidate Division SR1 and Gracilibacteria Code
There are other options as well, you can check the help for more details.
singularity exec mitohifi_master.sif mitohifi.py -hYou can submit this as a job script to run on HPC systems.
#!/bin/bash#SBATCH --nodes=1#SBATCH --ntasks=64#SBATCH --partition=<partition-name>#SBATCH --account=<account-name>#SBATCH --time=4-00:00:00#SBATCH --job-name=mitohifi_run#SBATCH --output=anvil-%x.%j.out#SBATCH --error=anvil-%x.%j.err#SBATCH --mail-user=${USER}@purdue.edu#SBATCH --mail-type=begin#SBATCH --mail-type=endml purge
singularity exec mitohifi_master.sif mitohifi.py \ -r maize-hifi.fasta \ -f maize/NC_007982.1.fasta \ -g maize/NC_007982.1.gb \ -t ${SLURM_CPUS_ON_NODE} \ -a plant \ -p 80 \ -o 13. Output
Section titled “3. Output”MitoHifi will produce a series of folders with the results. The main results will be in your working folder and they are:
final_mitogenome.fasta- the final mitochondria circularized and rotated to start at tRNA-Phefinal_mitogenome.gb- the final mitochondria annotated in GenBank format.final_mitogenome.coverage.png- the sequencing coverage throughout the final mitogenomefinal_mitogenome.annotation.png- the predicted genes throughout the final mitogenomecontigs_annotations.png- annotation plots for all potential contigscoverage_plot.png- reads coverage plot of filtered reads mapped to all potential contigscontigs_stats.tsv- containing the statistics of your assembled mitos such as the number of genes, size, whether it was circularized or not, if the sequence has frameshifts and etc…shared_genes.tsv- show comparison of annotation between close-related mitogenome and all potential contigs assembled
Here are the plots for the maize B73 mitochondrial genome:
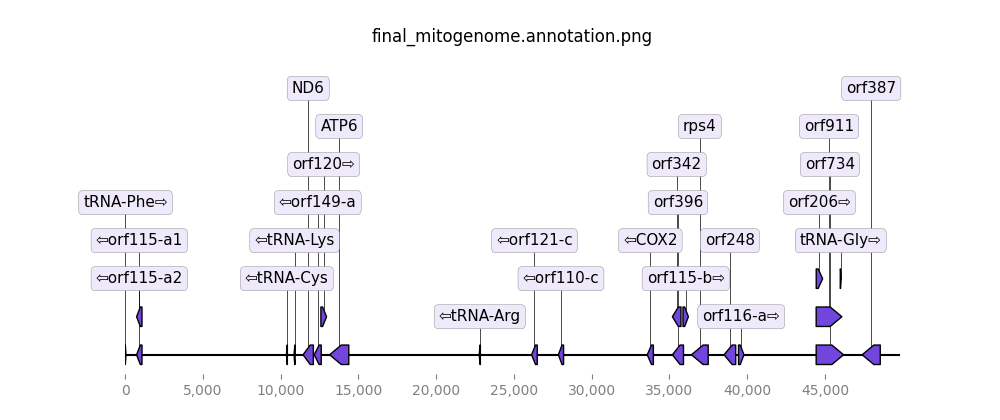
Figure 1: Predicted genes throughout the final mitogenome
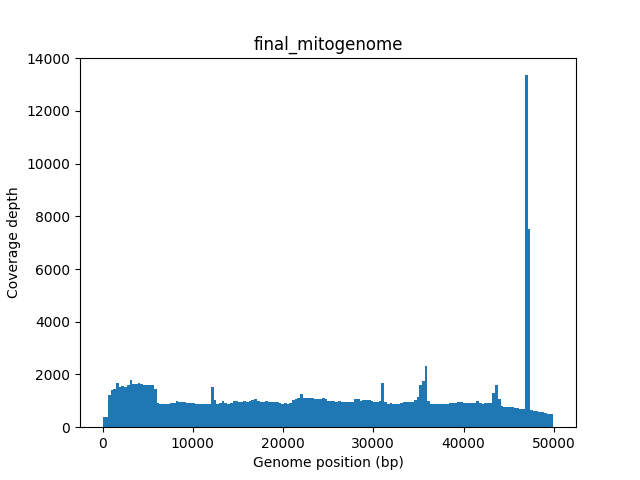
Figure 2: The sequencing coverage throughout the final mitogenome
4. References
Section titled “4. References”Uliano-Silva, M., Ferreira, J.G.R.N., Krasheninnikova, K. et al. MitoHiFi: a python pipeline for mitochondrial genome assembly from PacBio high fidelity reads. BMC Bioinformatics 24, 288 (2023). DOI: 10.1186/s12859-023-05385-y