Gene prediction using BRAKER3
BRAKER3 is a pipeline that combines GeneMark-ET and AUGUSTUS to predict genes in eukaryotic genomes. This pipeline is particularly useful for annotating newly sequenced genomes. The flexibility of BRAKER3 allows users to provide various input datasets for improving gene prediction accuracy. In this example, we will use various scenarios to predict genes in a Maize genome using BRAKER3. Following are the scenarios we will cover:
| Input Type | Case 1 | Case 2 | Case 3 | Case 4 | Case 5 | Case 6 | Case 7 | Case 8 |
|---|---|---|---|---|---|---|---|---|
| Genome | ✔️ | ✔️ | ✔️ | ✔️ | ✔️ | ✔️ | ✔️ | ✔️ |
| RNA-Seq | ❌ | ✔️^* | ✔️ | ❌ | ✔️ | ❌ | ❌ | ✔️ |
| Iso-Seq | ❌ | ❌ | ❌ | ❌ | ❌ | ❌ | ✔️ | ✔️ |
| Conserved proteins | ❌ | ❌ | ❌ | ✔️ | ✔️ | ❌ | ✔️ | ✔️ |
| Pretrained species model | ❌ | ❌ | ❌ | ❌ | ❌ | ✔️ | ❌ | ❌ |
minimal RNA-Seq data (one library/one tissue)
Installation
Section titled “Installation”We will use the apptainer tool to build a Singularity container for BRAKER3. The Singularity container will contain all the necessary dependencies and tools required to run BRAKER3. To build the Singularity container, run the following command:
apptainer build --fakeroot braker3.sif docker://teambraker/braker3:latestThis will create a Singularity container named braker3.sif with BRAKER3 installed.
Settng up BRAKER3
Section titled “Settng up BRAKER3”Before running BRAKER3, we need to set up:
GeneMark-ES/ET/EP/ETPlicense key- The
AUGUSTUS_CONFIG_PATHconfiguration path
The license key for GeneMark-ES/ET/EP/ETP can be obtained from the GeneMark website. Once downloaded, you need to place it in your home directory:
tar xf gm_key_64.gzcp gm_key_64 ~/.gm_keyFor the AUGUSTUS_CONFIG_PATH, we need to copy the config directory from the Singularity container to the scratch directory. This is required because BRAKER3 needs to write to the config directory, and the Singularity container is read-only. To copy the config directory, run the following command:
apptainer exec braker3.sif cp -r /opt/Augustus/config ${RCAC_SCRATCH}/braker/augustus_configRunning BRAKER3
Section titled “Running BRAKER3”The paths to the following variables need to be set:
BRAKER_SIF="${RCAC_SCRATCH}/braker/braker3.sif"AUGUSTUS_CONFIG_PATH="${RCAC_SCRATCH}/braker/augustus_config"GENEMARK_PATH="/opt/ETP/bin/gmes"genome="${RCAC_SCRATCH}/braker/Zm-B73-REFERENCE-NAM-5.0_softmasked.fa"workdir=${PWD}/$(basename ${genome%.*})_brakerInput datasets
Section titled “Input datasets”With genome only (no external evidence)
| Input | Type |
|---|---|
| Genome | B73.v5 (softmasked) |
| RNA-Seq data | None |
| Protein sequences | None |
| Long-read data | None |
| Pretrained species model | None |
mkdir -p ${workdir}apptainer exec --bind ${RCAC_SCRATCH} ${BRAKER_SIF} braker.pl \ --AUGUSTUS_CONFIG_PATH=${AUGUSTUS_CONFIG_PATH} \ --GENEMARK_PATH=${GENEMARK_PATH} \ --esmode \ --genome=${genome} \ --species=Zm_$(date +"%Y%m%d").c1 \ --workingdir=${workdir} \ --gff3 \ --threads ${SLURM_CPUS_ON_NODE}with minimal RNA-Seq data (one library/one tissue)
| Input | Type |
|---|---|
| Genome | B73.v5 (softmasked) |
| RNA-Seq data | RNAseq (single library) |
| Protein sequences | None |
| Long-read data | None |
| Pretrained species model | None |
mkdir -p ${workdir}apptainer exec --bind ${RCAC_SCRATCH} ${BRAKER_SIF} braker.pl \ --AUGUSTUS_CONFIG_PATH=${AUGUSTUS_CONFIG_PATH} \ --GENEMARK_PATH=${GENEMARK_PATH} \ --rnaseq_sets_ids=B73_V11_middle_MN01042 \ --rnaseq_sets_dirs=${RCAC_SCRATCH}/RNAseq/ \ --genome=${genome} \ --species=Zm_$(date +"%Y%m%d").c2 \ --workingdir=${workdir} \ --gff3 \ --threads ${SLURM_CPUS_ON_NODE}with exhaustive RNA-Seq data (11 tissues)
| Input | Type |
|---|---|
| Genome | B73.v5 (softmasked) |
| RNA-Seq data | RNAseq (11 tissues) |
| Protein sequences | None |
| Long-read data | None |
| Pretrained species model | None |
rna_seq_sets_ids="B73_8DAS_root_MN01011,B73_8DAS_root_MN01012,B73_8DAS_shoot_MN01021,B73_8DAS_shoot_MN01022,B73_16DAP_embryo_MN01101,B73_16DAP_embryo_MN01102,B73_16DAP_endosperm_MN01091,B73_16DAP_endosperm_MN01092,,B73_R1_anther_MN01081,B73_R1_anther_MN01082,B73_R1_anther_MNA1081,B73_V11_base_MN01031,B73_V11_base_MN01032,B73_V11_middle_MN01041,B73_V11_middle_MN01042,B73_V11_middle_MN01043,B73_V11_tip_MN01051,B73_V11_tip_MN01052,B73_V18_ear_MN01071,B73_V18_ear_MN01072,B73_V18_tassel_MN01061,B73_V18_tassel_MN01062"mkdir -p ${workdir}apptainer exec --bind ${RCAC_SCRATCH} ${BRAKER_SIF} braker.pl \ --AUGUSTUS_CONFIG_PATH=${AUGUSTUS_CONFIG_PATH} \ --GENEMARK_PATH=${GENEMARK_PATH} \ --rnaseq_sets_ids=${rnaseq_sets_ids} \ --rnaseq_sets_dirs=${RCAC_SCRATCH}/rnaseq/ \ --genome=${genome} \ --species=Zm_$(date +"%Y%m%d").c3 \ --workingdir=${workdir} \ --gff3 \ --threads ${SLURM_CPUS_ON_NODE}with conserved protein sequences
| Input | Type |
|---|---|
| Genome | B73.v5 (softmasked) |
| RNA-Seq data | None |
| Protein sequences | Viridiplantae protein sequences |
| Long-read data | None |
| Pretrained species model | None |
Using the orthodb-clades tool, we can download protein sequences for a specific clade. In this scenario, since we are using the Maize genome, we can download the clade specific Viridiplantae.fa OrthoDB v12 protein sets.
git clone git@github.com:tomasbruna/orthodb-clades.gitml biocontainersml snakemakesnakemake --cores ${SLURM_CPUS_ON_NODE}When this is done, you should see a folder named clade with Viridiplantae.fa in the orthodb-clades directory. We will use this as one of the input datasets for BRAKER3. The following command will run BRAKER3 with the input genome and protein sequences:
protein_sequences="${RCAC_SCRATCH}/braker/orthodb-clades/clade/Viridiplantae.fa"apptainer exec --bind ${RCAC_SCRATCH} ${BRAKER_SIF} braker.pl \ --AUGUSTUS_CONFIG_PATH=${AUGUSTUS_CONFIG_PATH} \ --GENEMARK_PATH=${GENEMARK_PATH} \ --species=Zm_$(date +"%Y%m%d").1a \ --prot_seq=${proteins} \ --genome=${genome} \ --species=Zm_$(date +"%Y%m%d").c4 \ --workingdir=${workdir} \ --gff3 \ --threads ${SLURM_CPUS_ON_NODE}with RNA-Seq and conserved protein sequences
| Input | Type |
|---|---|
| Genome | B73.v5 (softmasked) |
| RNA-Seq data | RNAseq (11 tissues) |
| Protein sequences | Viridiplantae protein sequences |
| Long-read data | None |
| Pretrained species model | None |
mkdir -p ${workdir}apptainer exec --bind ${RCAC_SCRATCH} ${BRAKER_SIF} braker.pl \ --AUGUSTUS_CONFIG_PATH=${AUGUSTUS_CONFIG_PATH} \ --GENEMARK_PATH=${GENEMARK_PATH} \ --rnaseq_sets_ids=${rnaseq_sets_ids} \ --rnaseq_sets_dirs=${RCAC_SCRATCH}/rnaseq/ \ --prot_seq=${proteins} \ --genome=${genome} \ --species=Zm_$(date +"%Y%m%d").c5 \ --workingdir=${workdir} \ --gff3 \ --threads ${SLURM_CPUS_ON_NODE}with pretrained species model (“Maize”)
| Input | Type |
|---|---|
| Genome | B73.v5 (softmasked) |
| RNA-Seq data | None |
| Protein sequences | None |
| Long-read data | None |
| Pretrained species model | ”Maize” |
mkdir -p ${workdir}apptainer exec --bind ${RCAC_SCRATCH} ${BRAKER_SIF} braker.pl \ --AUGUSTUS_CONFIG_PATH=${AUGUSTUS_CONFIG_PATH} \ --GENEMARK_PATH=${GENEMARK_PATH} \ --skipAllTraining \ --genome=${genome} \ --species=maize5 \ --workingdir=${workdir} \ --gff3 \ --threads ${SLURM_CPUS_ON_NODE}with Iso-Seq and conserved protein sequences
| Input | Type |
|---|---|
| Genome | B73.v5 (softmasked) |
| RNA-Seq data | None |
| Protein sequences | Viridiplantae protein sequences |
| Long-read data | Iso-Seq data |
| Pretrained species model | None |
The IsoSeq data for maize (B73) was obtained from the publication PMC7028979 and is available in the ENA BioProject PRJEB32007.
To proceed, you will need the original files listed in the Submitted files: FTP column of the BioProject page. We will download the data (.bam files) and process them using the isoseq3 tool to demultiplex and map the reads to the B73 reference genome.
The primers and adapters required for demultiplexing were sourced from the original publication (Supplementary Table 1).
ml purgeml anacondaconda activate isoseqfor input in *subreads.bam; dobase=$(basename ${input} |sed 's/.subreads.bam//g')# convert subreads to ccsccs ${input} ${base}.ccs.bam --skip-polish --min-passes 1# demultiplexlima ${base}.ccs.bam primer.fasta ${base}_lima.bam --isoseq --dump-clipsdone# move the files to separate directoriesfor f in B73 Ki11 Ki11xB73 B73xKi11; domkdir -p $f;mv *_${f}_* ./${f}/;donecd B73# merge the filesls *.bam > input.fofnml purgeml biocontainersml bamtools samtoolsbamtools merge -list input.fofn -out merged_B73.bam# convert bam to fastqsamtools fastq --threads ${SLURM_CPUS_ON_NODE} -o merged_B73.fastq merged_B73.bamml minimap2minimap2 \ -t ${SLURM_CPUS_ON_NODE}\ -ax splice:hq \ -uf ${genome} \ merged_B73.fastq > isoseq_B73.samsamtools view \ -bS \ --threads ${threads} \ -o isoseq.bam \ isoseq_b73.sam \samtools sort \ --threads ${threads} \ -o isoseq_sorted.bam \ isoseq.bamWe will need isoseq_sorted.bam (and merged_B73.fastq) for the case 8 as well.
For this case 7, we only need isoseq_sorted.bam. To setup BRAKER3 with the Iso-Seq data and conserved protein sequences:
isoseq="${RCAC_SCRATCH}/isoseq/isoseq_sorted.bam"apptainer exec --bind ${RCAC_SCRATCH} ${BRAKER_SIF} braker.pl \ --AUGUSTUS_CONFIG_PATH=${AUGUSTUS_CONFIG_PATH} \ --GENEMARK_PATH=${GENEMARK_PATH} \ --prot_seq=${proteins} \ –-bam=${isoseq} \ --genome=${genome} \ --species=Zm_$(date +"%Y%m%d").c7 \ --workingdir=${workdir} \ --gff3 \ --threads ${SLURM_CPUS_ON_NODE}with Iso-Seq, RNA-Seq and conserved protein sequences
| Input | Type |
|---|---|
| Genome | B73.v5 (softmasked) |
| RNA-Seq data | RNAseq (11 tissues) |
| Protein sequences | Viridiplantae protein sequences |
| Long-read data | Iso-Seq data |
| Pretrained species model | None |
To run this, you need to first run case 3 (full-RNAseq data) [BRAKER-1] and case 5 (conserved proteins data) [BRAKER-2]. You will also need the Iso-Seq BAM file generated in case 7.
The steps are as follows:
- Run
BRAKERusing the spliced alignments of short-read RNA-seq (here case 3 with full-RNAseq data). - Run
BRAKERusing the conserved proteins data (here case 5 with conserved proteins data). - Run
GeneMarkS-Tprotocol on the Iso-Seq data to predict protein-coding regions in the transcripts:- map the long reads to the genome using minimap2 (here case 7
isoseq_sorted.bam) - collapse redundant isoforms
- predict protein-coding regions using
GeneMarkS-T
- map the long reads to the genome using minimap2 (here case 7
- Run the long read version of
TSEBRAto combine the three gene sets using all extrinsic evidence
Since we have already run case 3 and case 5, we will proceed with the remaining steps.
collapse_isoforms_by_sam.py \ --input merged.fastq \ --fq \ -b isoseq_sorted.bam \ -o isoseq_sorted \ --dun-merge-5-shorter \ --cpus ${SLURM_CPUS_ON_NODE}stringtie2fa.py \ -g ${genome} \ -f isoseq_sorted.collapsed.gff \ -o cupcake.fa
gmst.pl \ --strand direct \ cupcake.fa.mrna \ --output gmst.out \ --format GFFgit clone https://github.com/Gaius-Augustus/BRAKERcd BRAKER && \ git checkout long-reads && \ cd ..BRAKER/scripts/gmst2globalCoords.py \ -t isoseq_sorted.collapsed.gff \ -p gmst.out \ -o gmst.global.gtf \ -g ${genome}We will need the hintsfile.gff and augustus.hints.gtf files from case 3 and case 5.
We will also need the gmst.global.gtf file generated from the GeneMarkS-T protocol.
The following command will run TSEBRA to combine the three gene sets:
ln -s ${RCAC_SCRATCH}/braker/case3/Augustus/augustus.hints.gtf braker1.augustus.hints.gtfln -s ${RCAC_SCRATCH}/braker/case5/Augustus/augustus.hints.gtf braker2.augustus.hints.gtfln -s ${RCAC_SCRATCH}/braker/case3/hintsfile.gff braker1.hintsfile.gffln -s ${RCAC_SCRATCH}/braker/case5/hintsfile.gff braker2.hintsfile.gffln -s ${RCAC_SCRATCH}/braker/case7/genemark_st/gmst.global.gtfgit clone https://github.com/Gaius-Augustus/TSEBRAcd TSEBRAgit checkout long-readsapptainer exec --bind ${RCAC_SCRATCH} ${BRAKER_SIF} ./TSEBRA/bin/tsebra.py \ -g braker1.augustus.hints.gtf,braker2.augustus.hints.gtf \ -e braker1.hintsfile.gff,braker2.hintsfile.gff \ -l gmst.global.gtf \ -c ./TSEBRA/config/long_reads.cfg \ -o tsebra.gtfml purgeml biocontainersml cufflinksgffread tsebra.gtf \ -g ${genome} \ -y tsebra_pep.fa \ -x tsebra_cds.faProcessing output
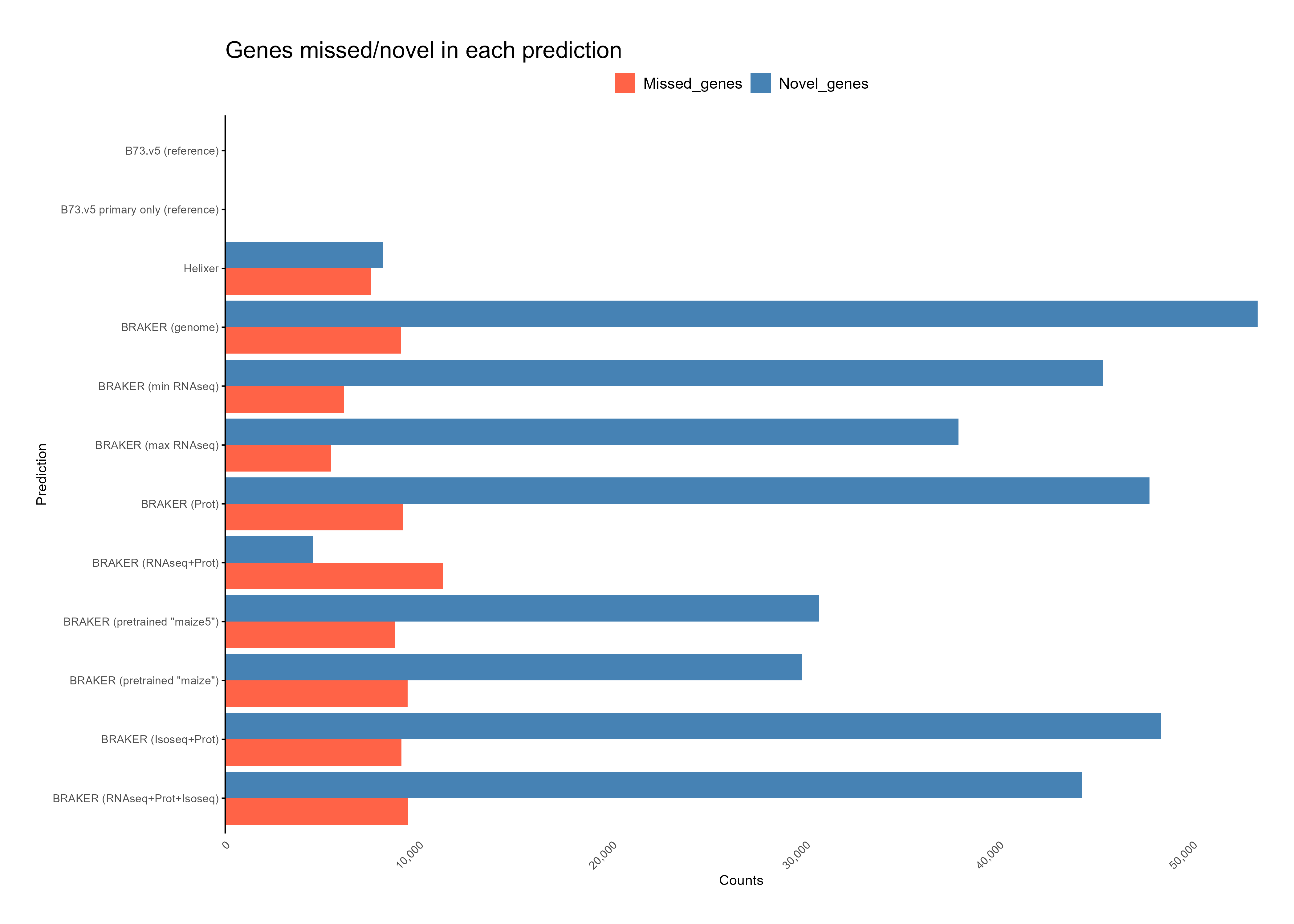
Section titled “Processing output”Comparing and Evaluating
Section titled “Comparing and Evaluating”A. BUSCO profiling
Section titled “A. BUSCO profiling”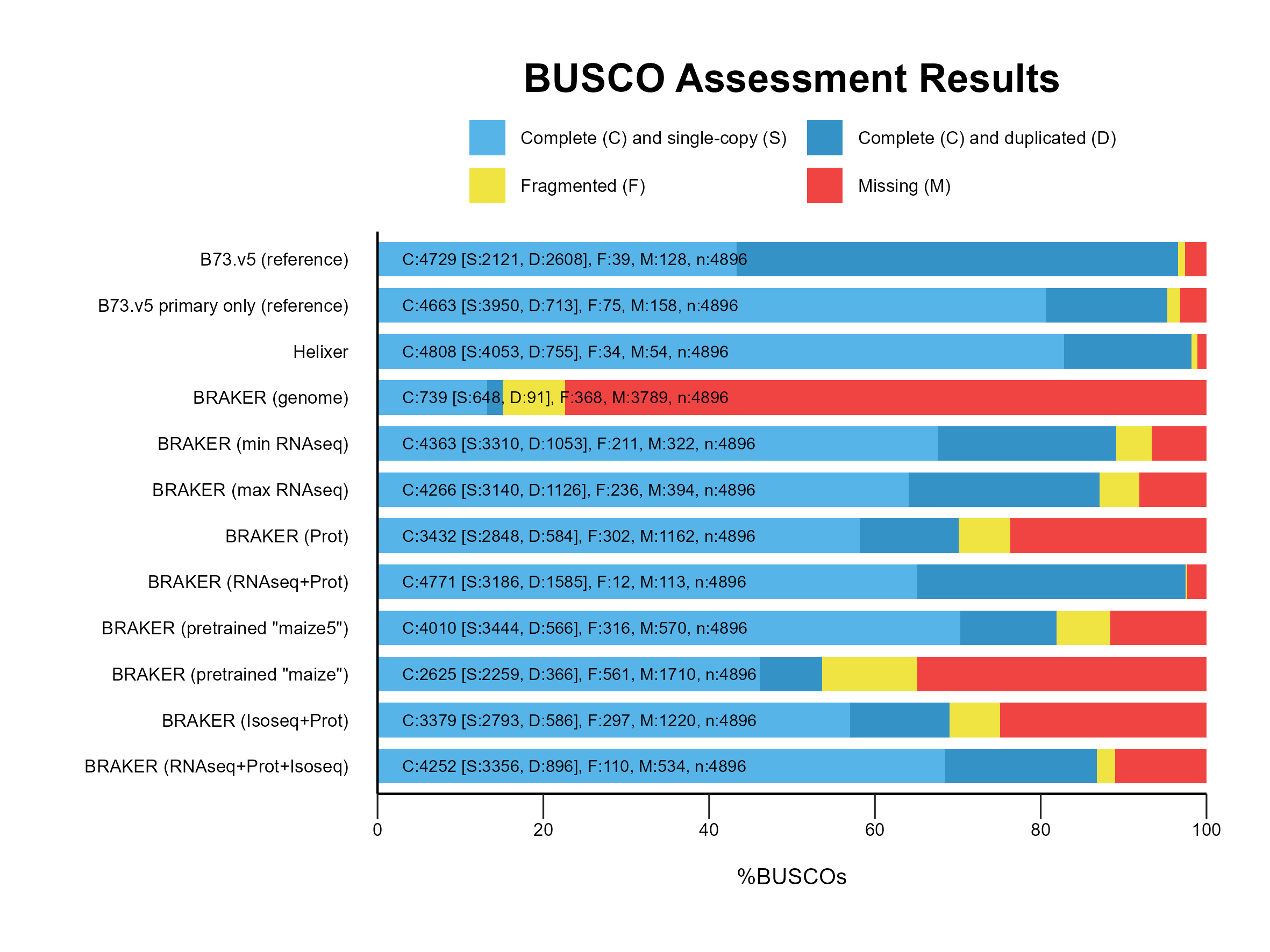
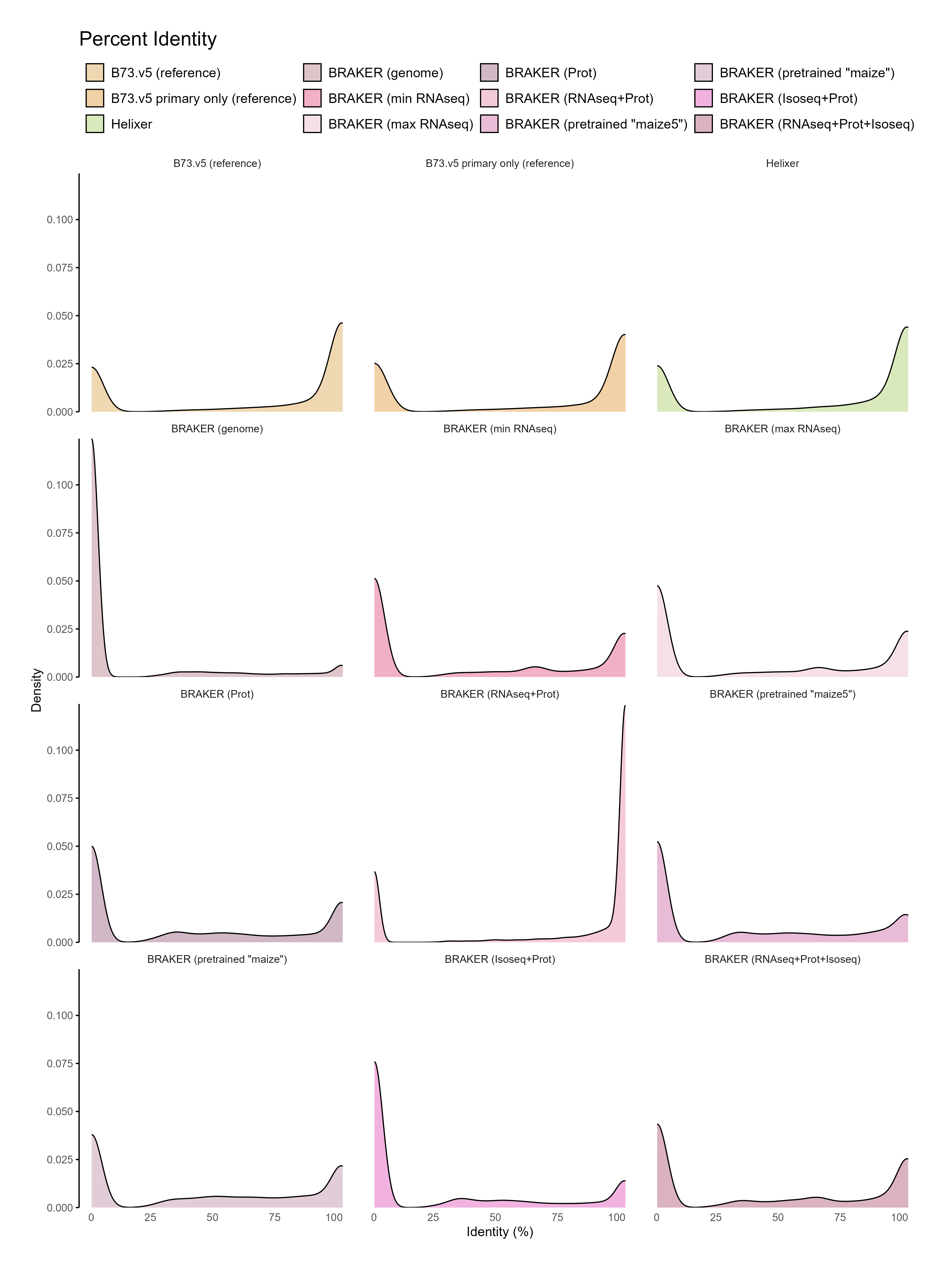
B. Reference comparison
Section titled “B. Reference comparison”With Isoforms
Section titled “With Isoforms”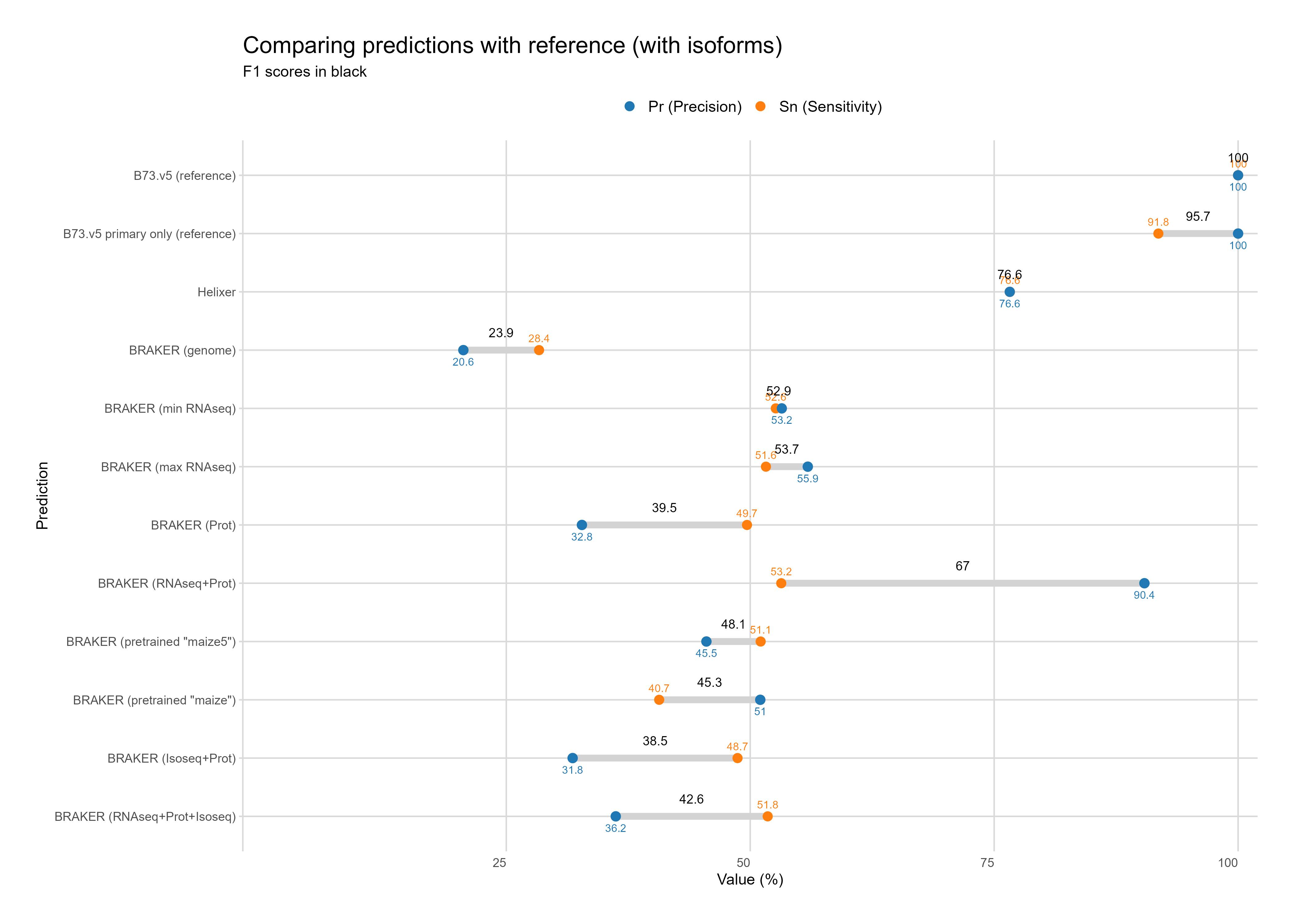
Without Isoforms
Section titled “Without Isoforms”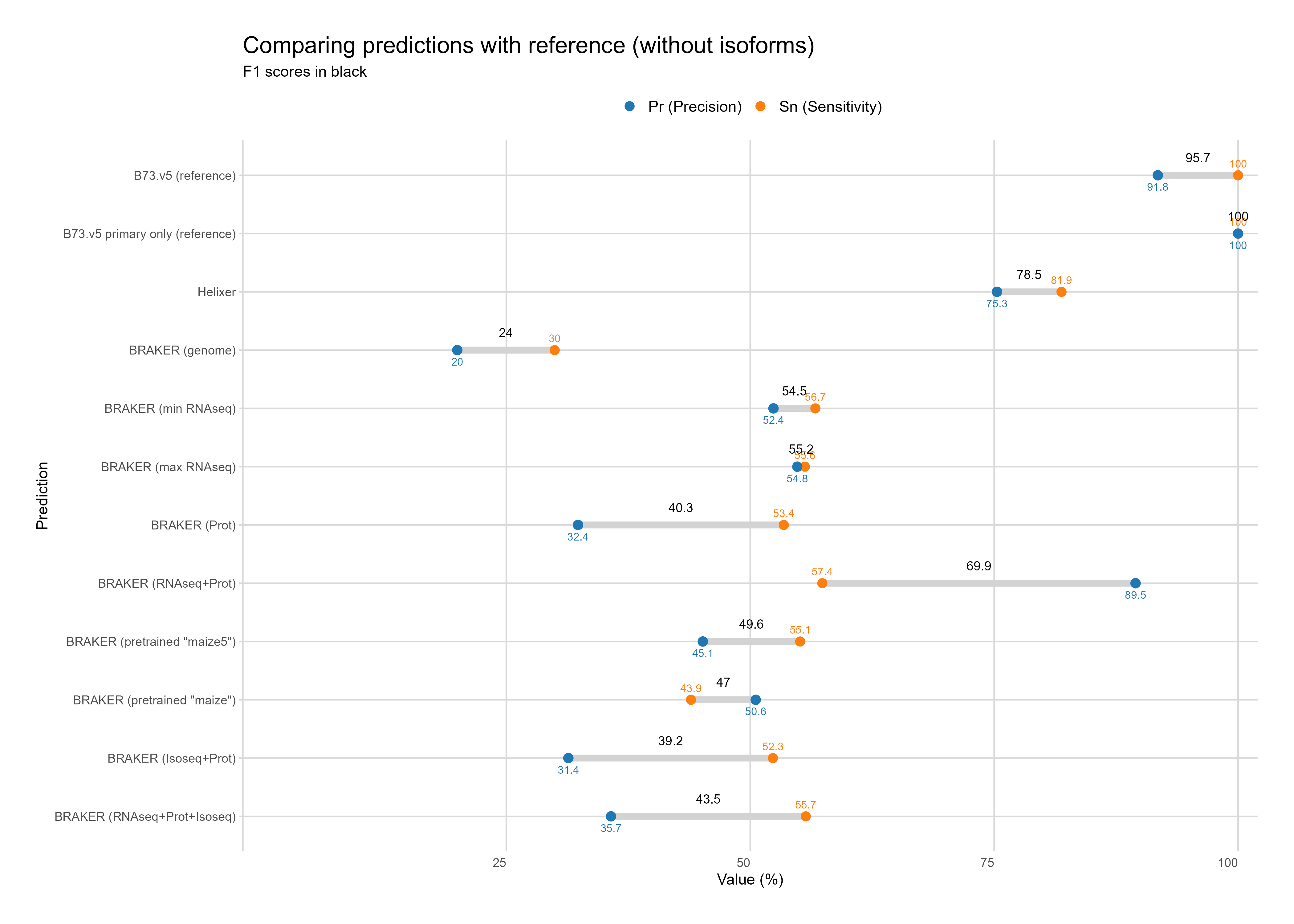
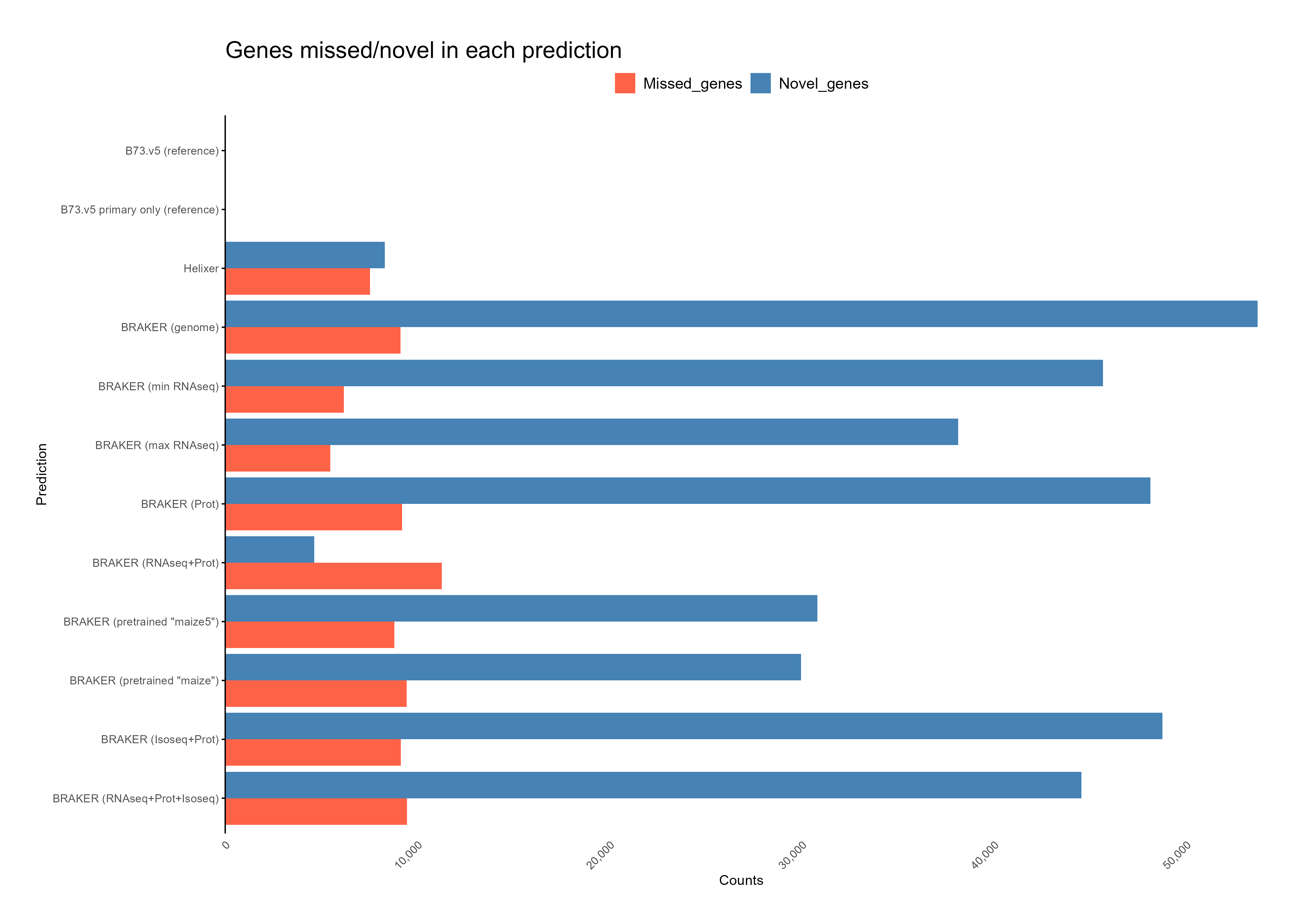
C. Feature assignment
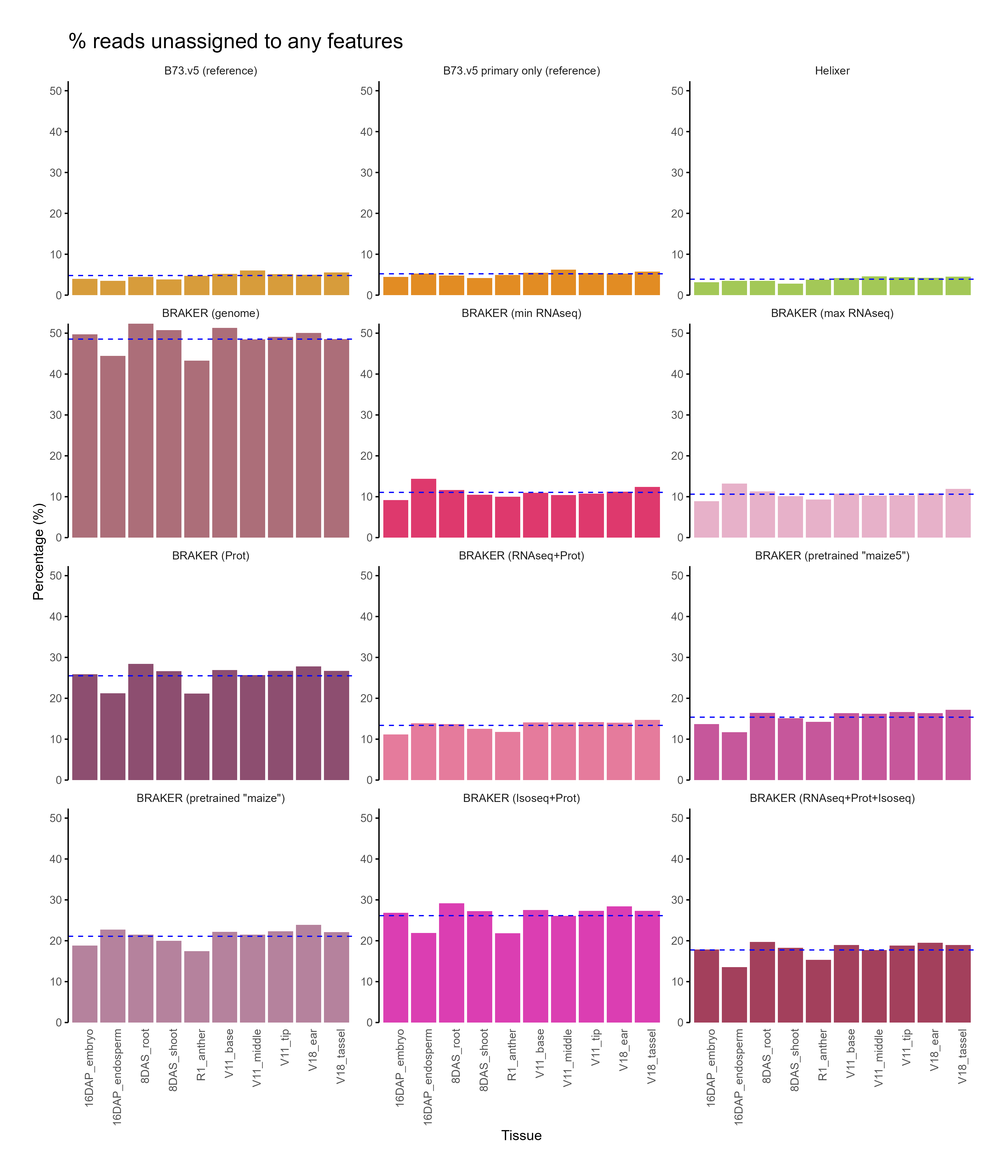
Section titled “C. Feature assignment”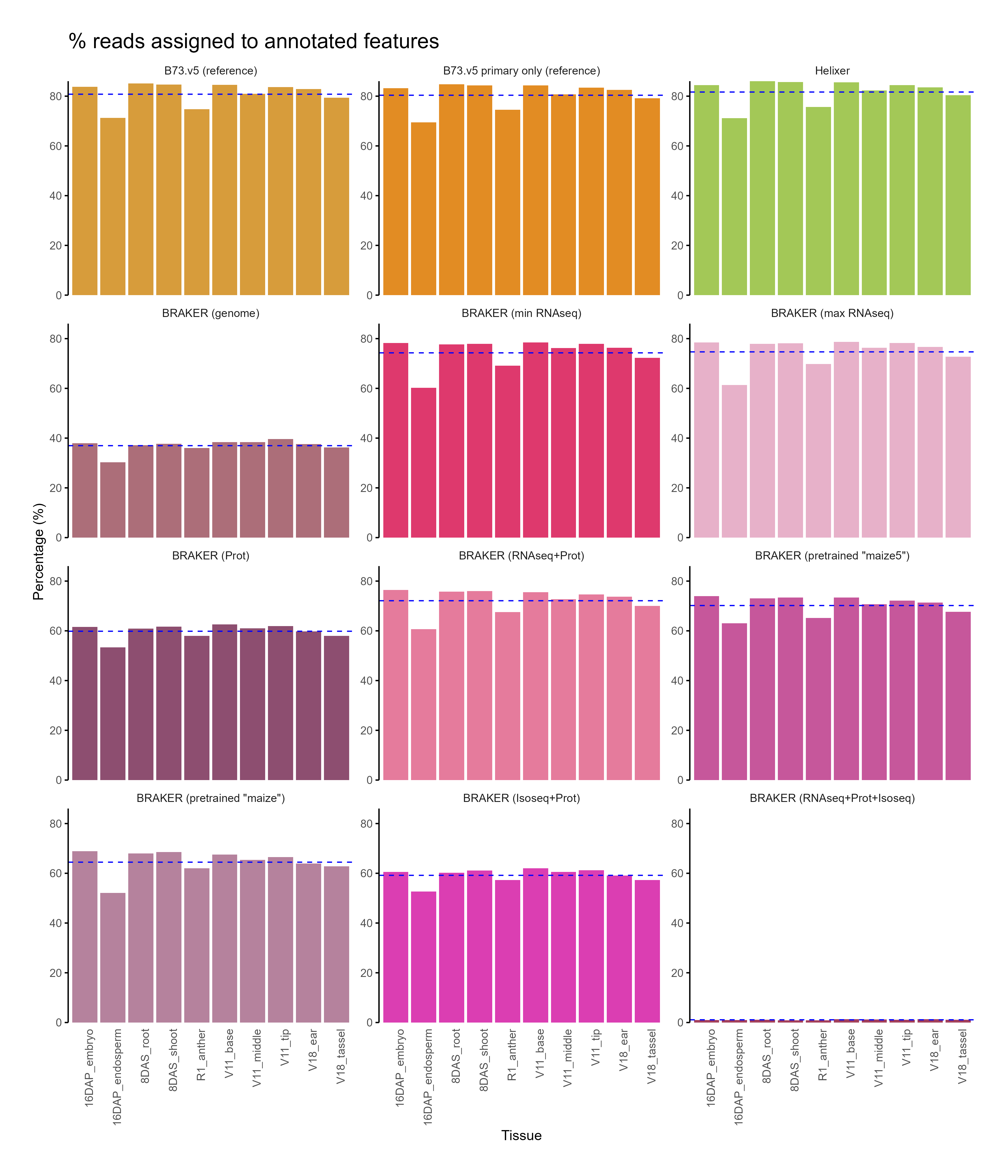
D. Functional annotation
Section titled “D. Functional annotation”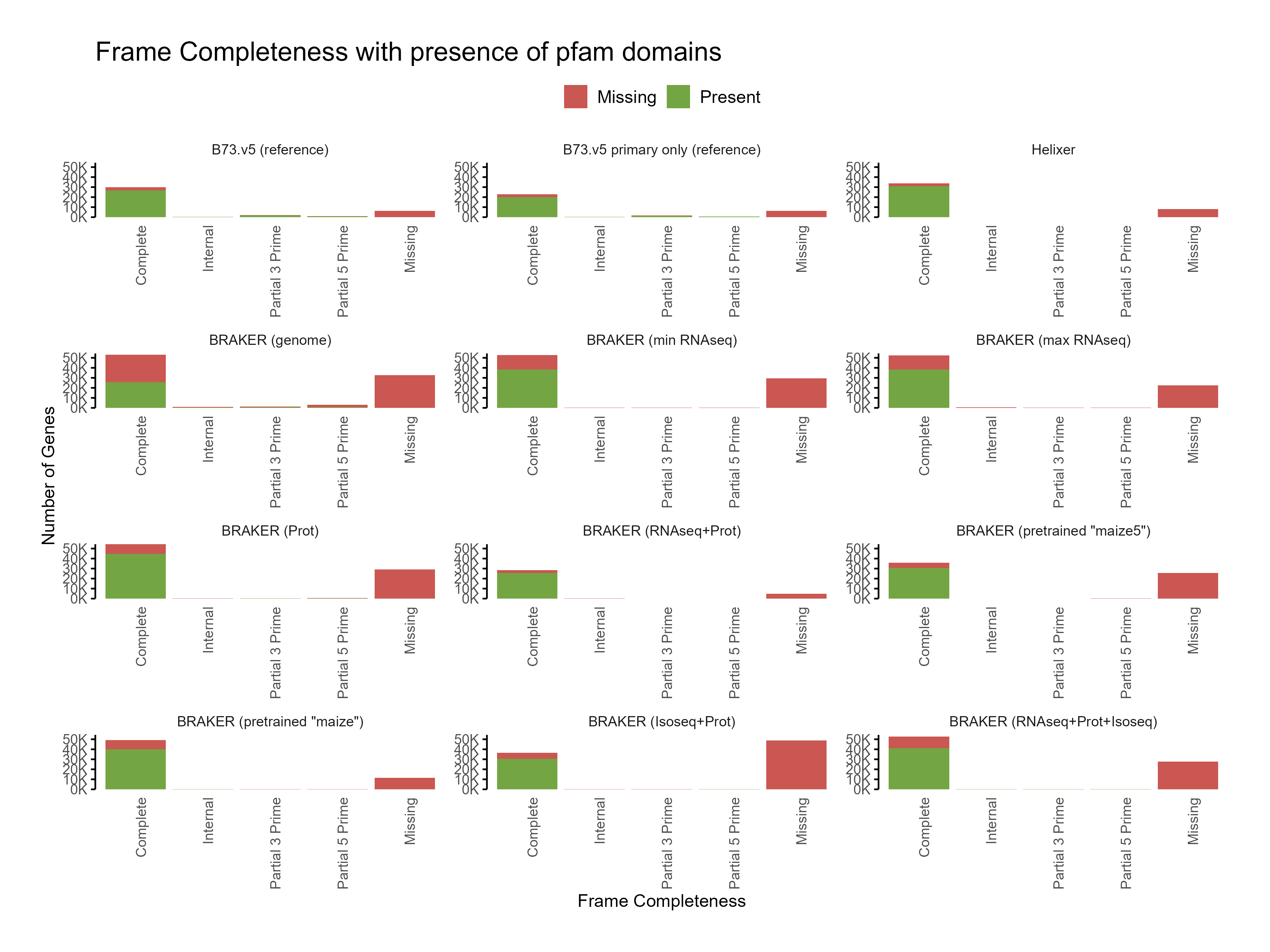
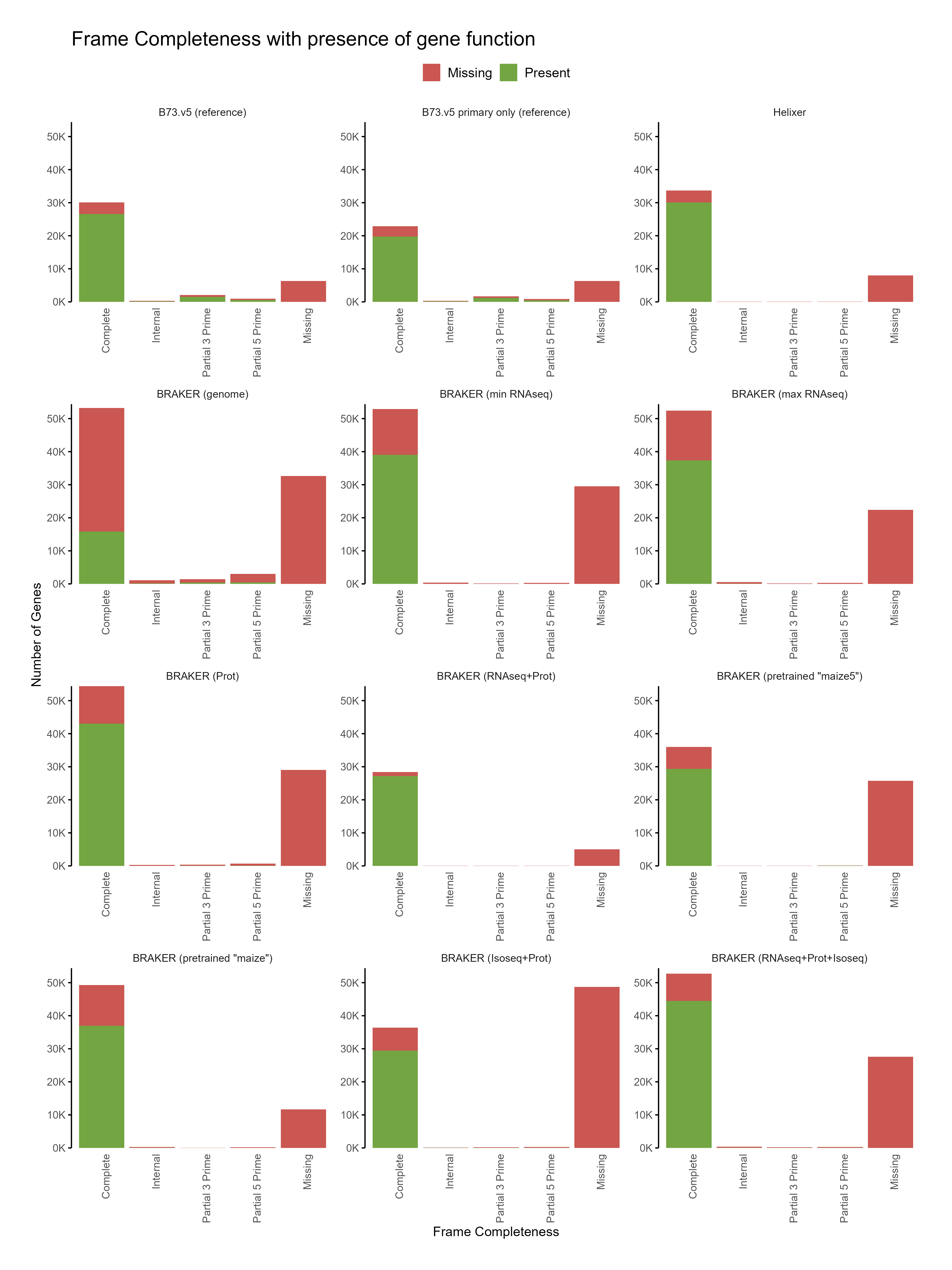
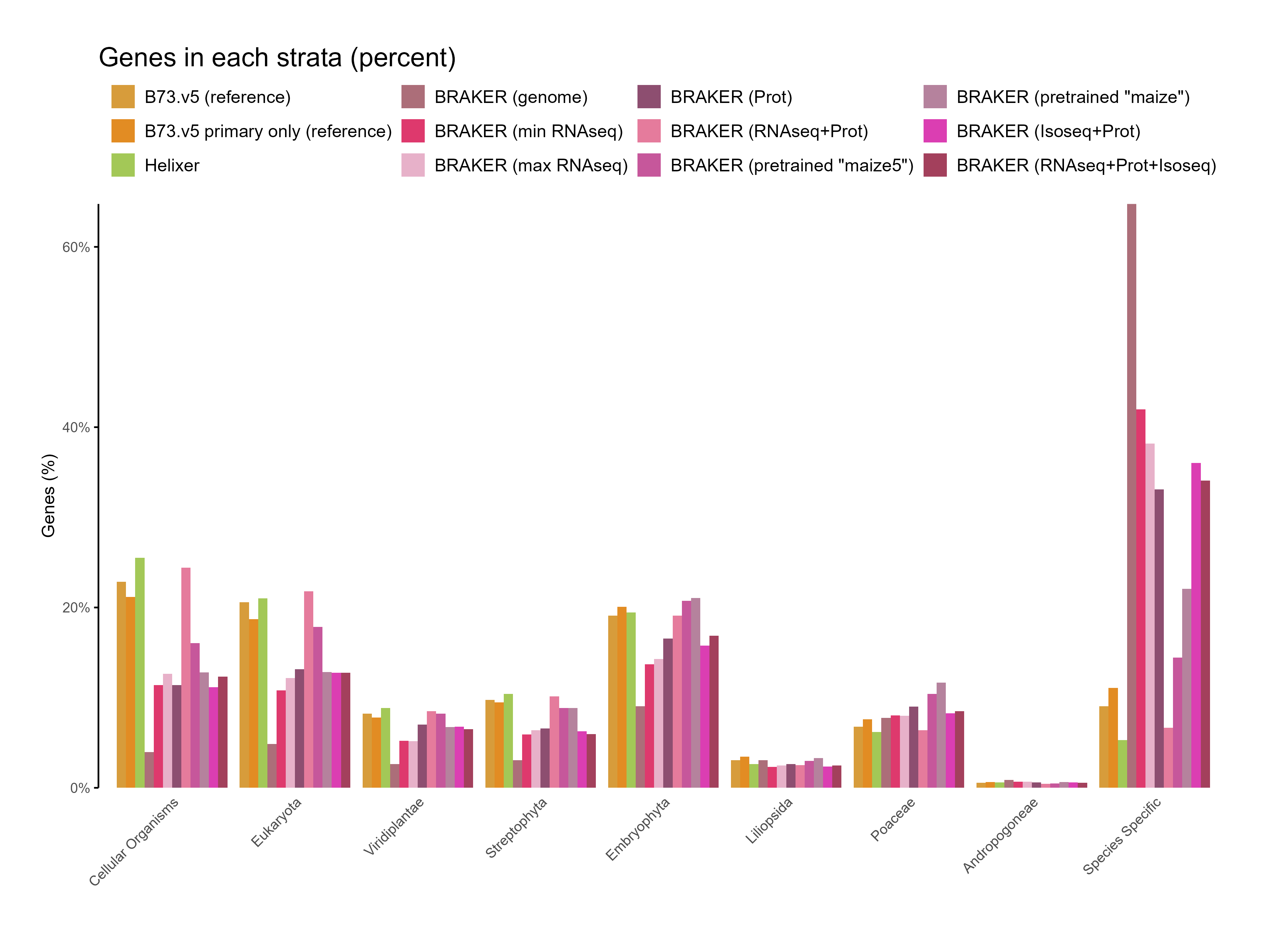
E. Phylostrata analysis
Section titled “E. Phylostrata analysis”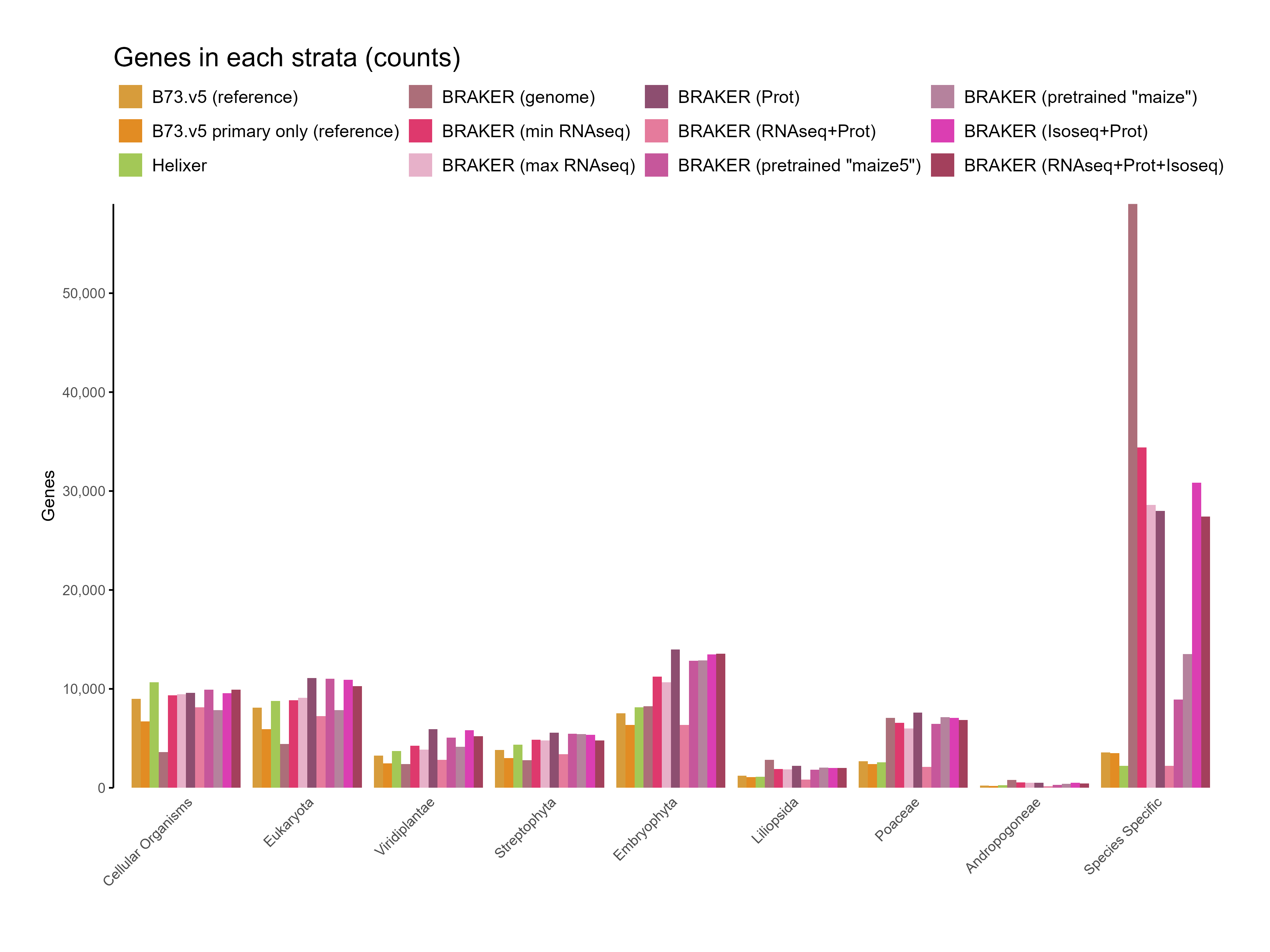
F. GFF3 stats
Section titled “F. GFF3 stats”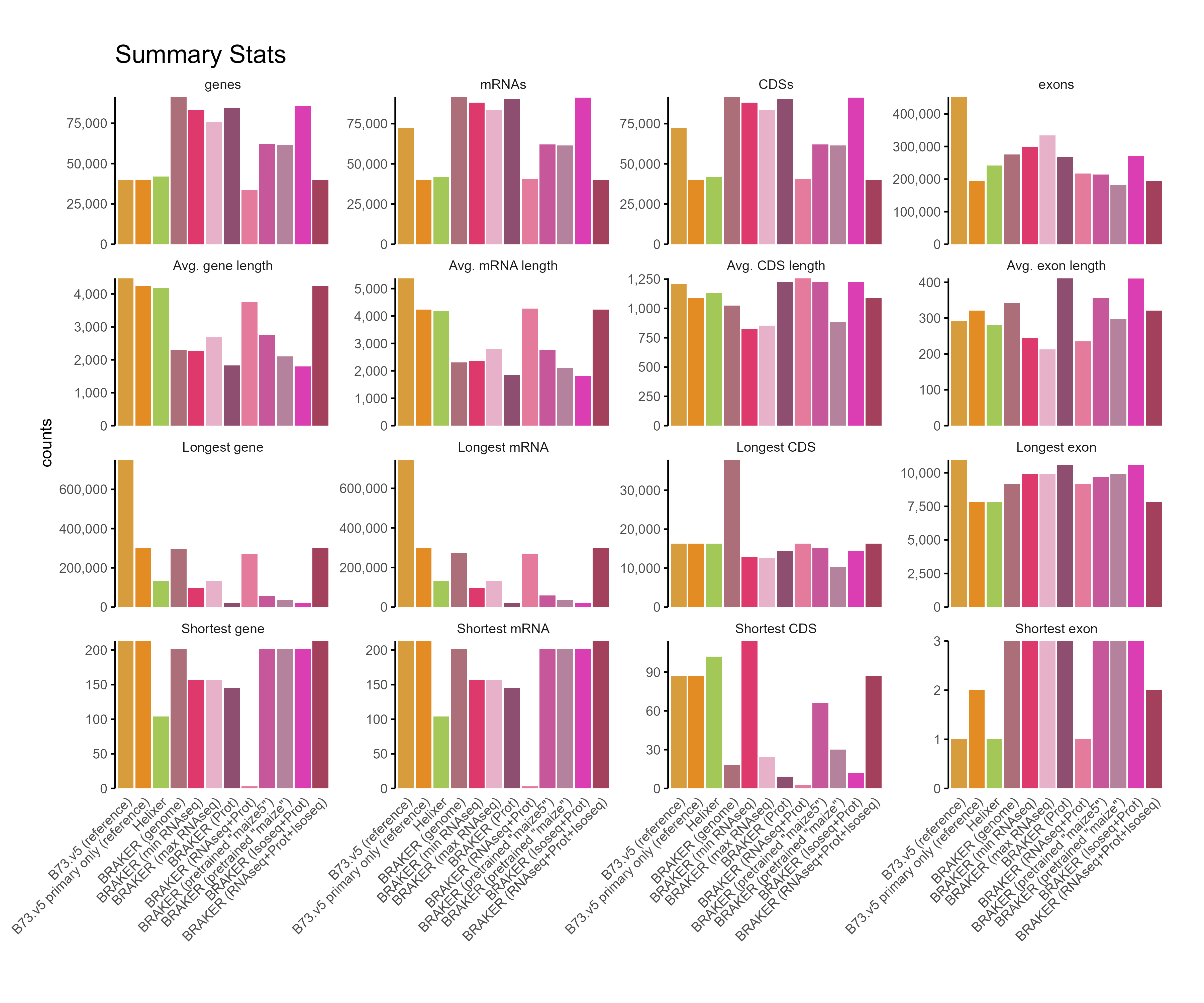
G. OMArk assesment
Section titled “G. OMArk assesment”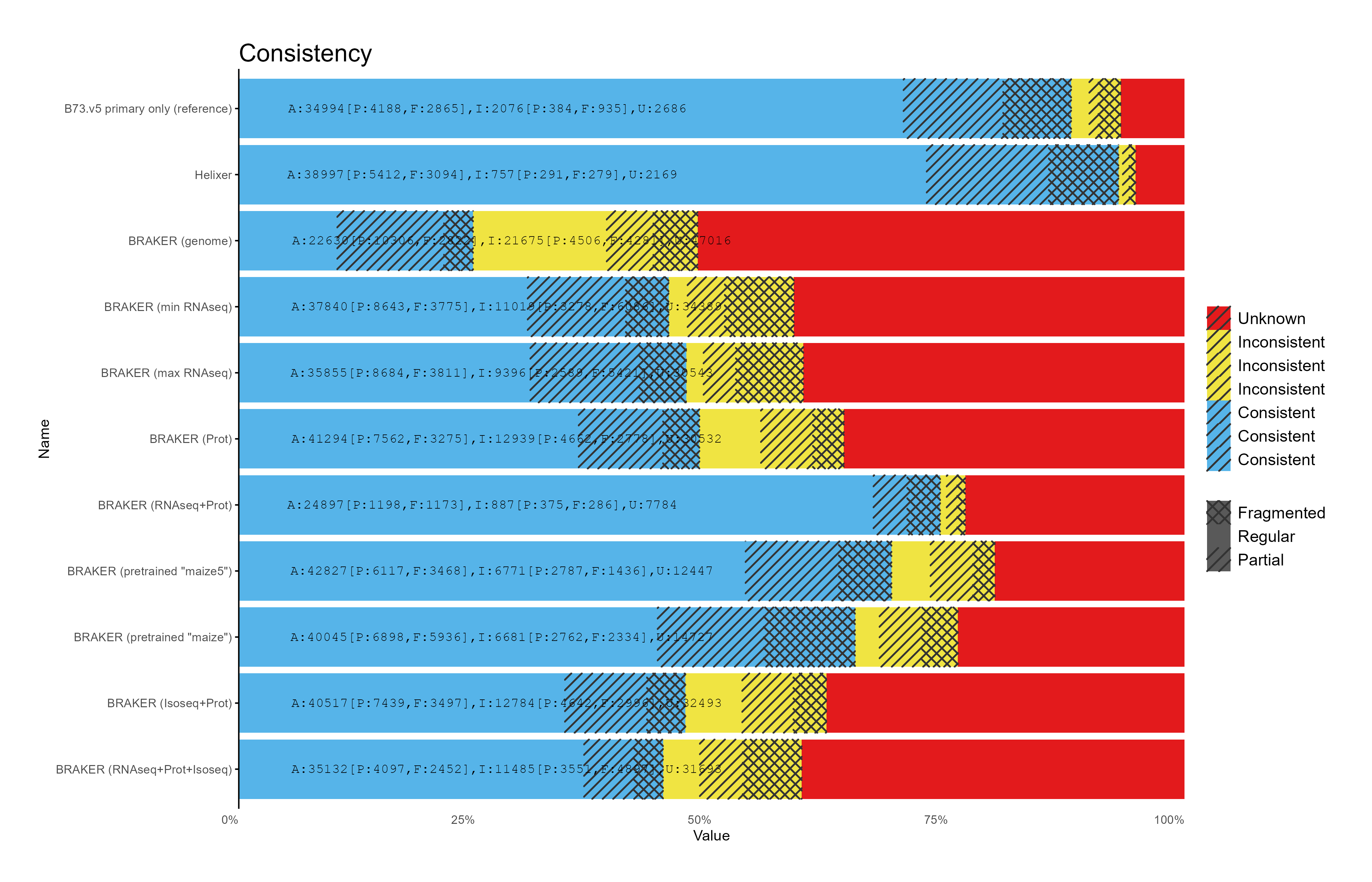
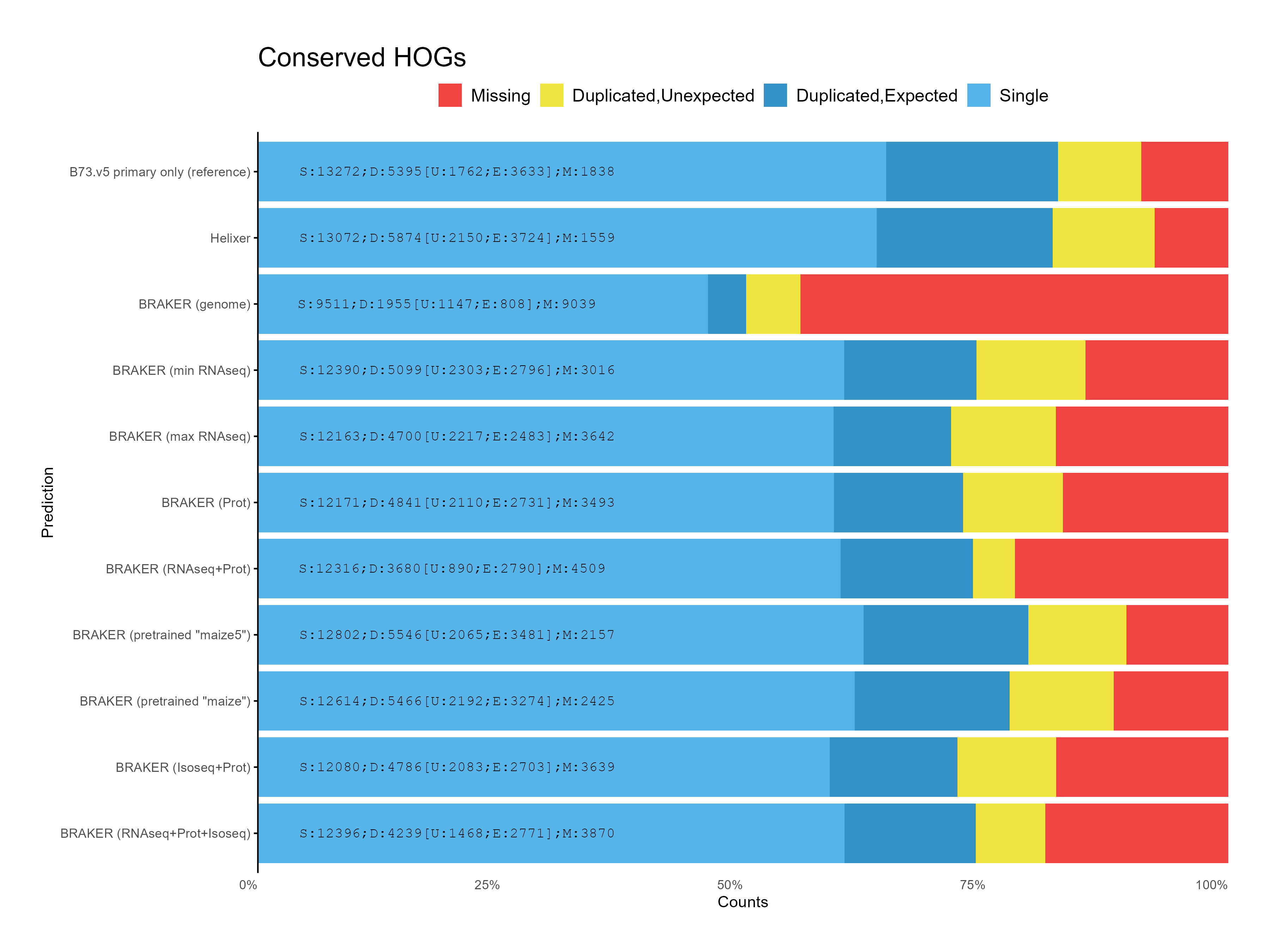
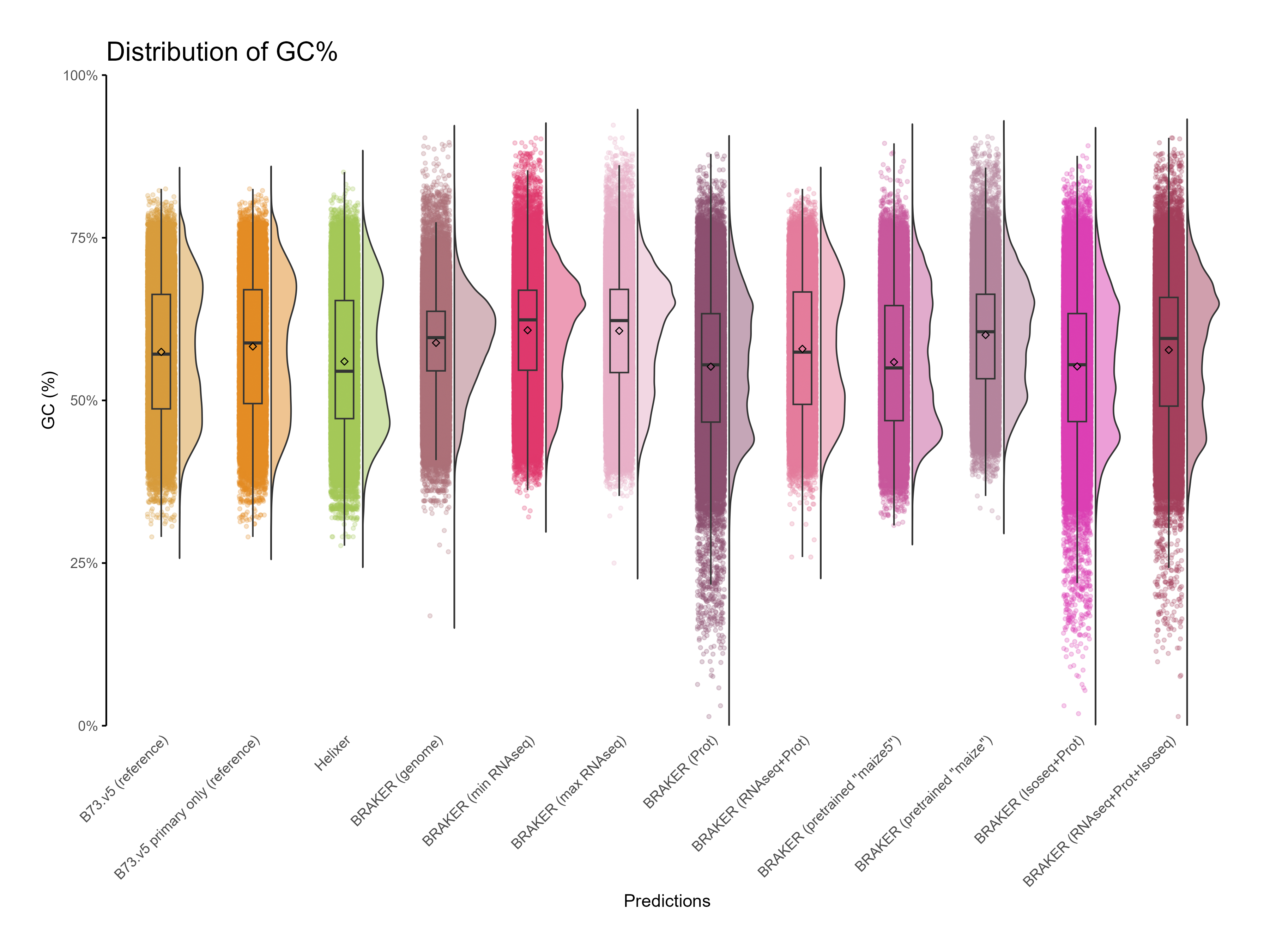
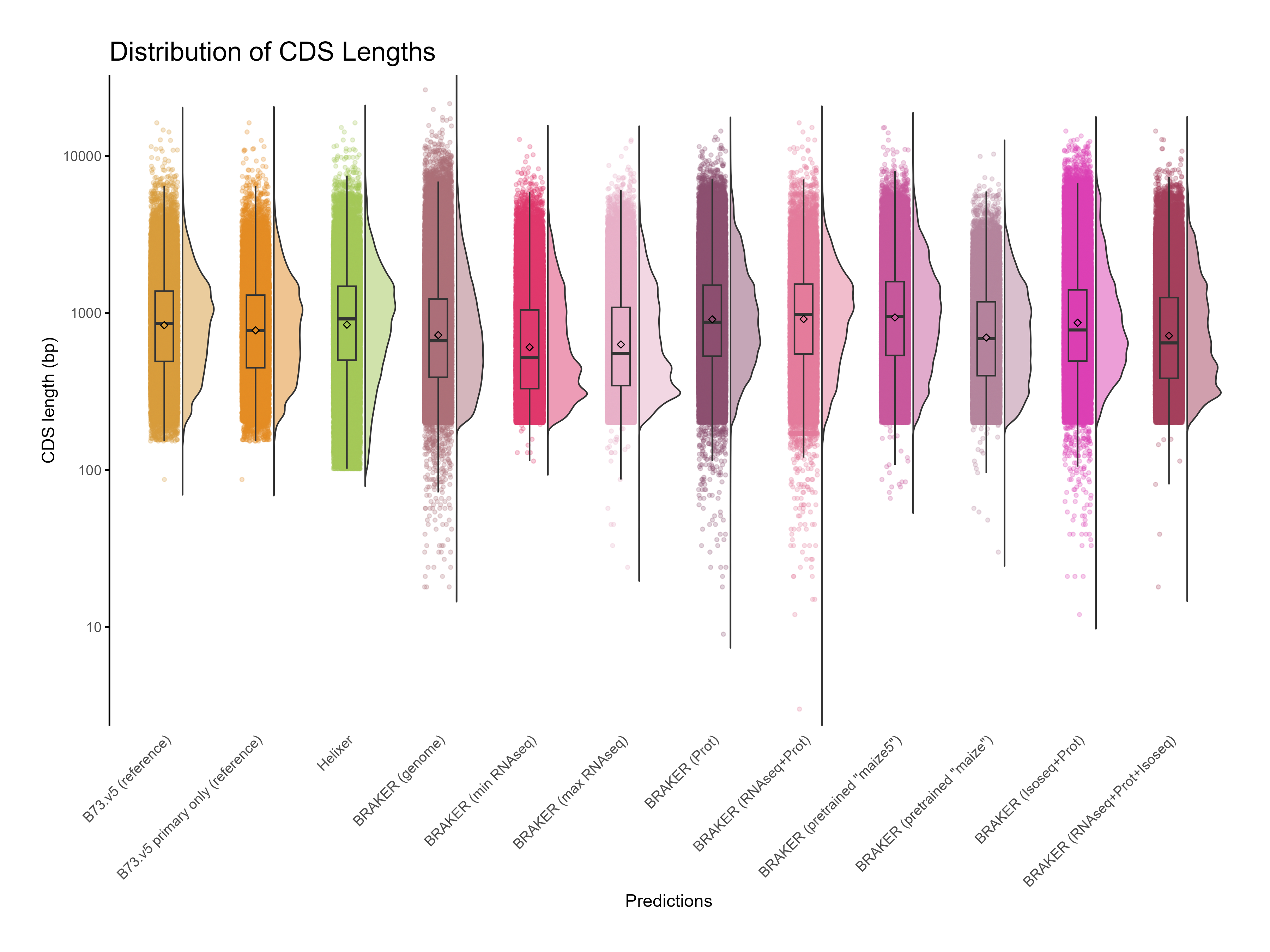
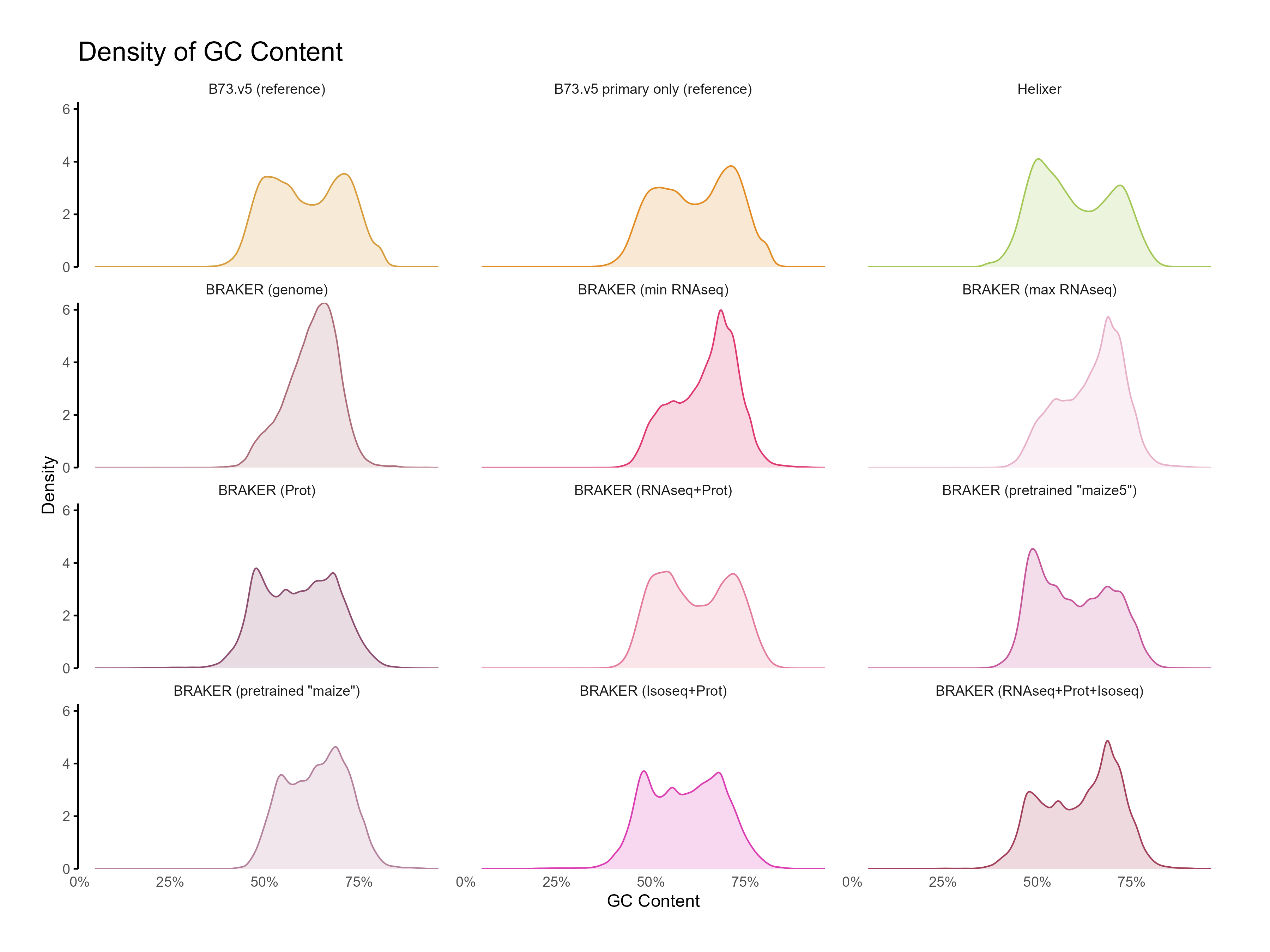
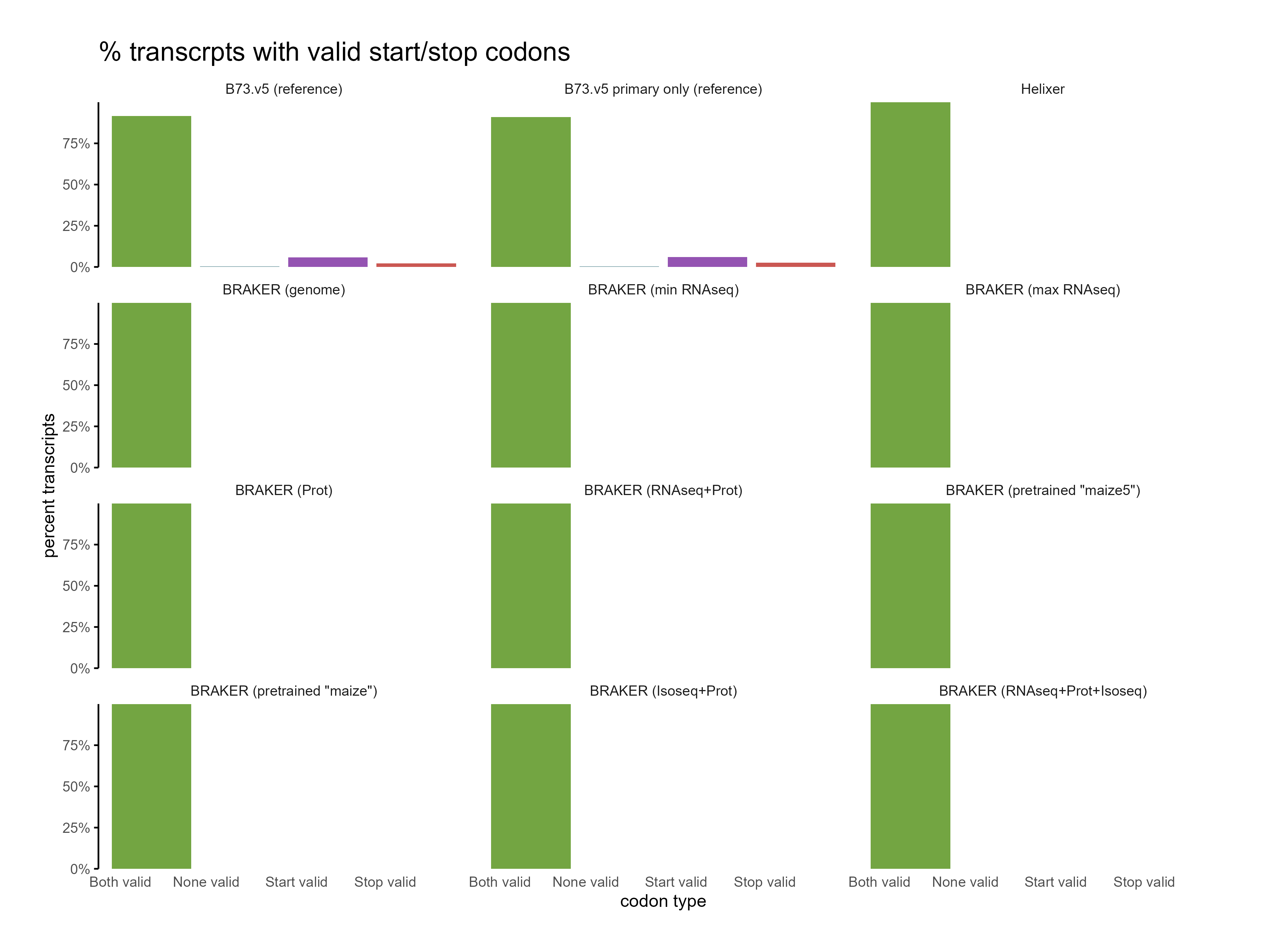
H. CDS assesments
Section titled “H. CDS assesments”