Gene prediction using Helixer
Helixer is a deep learning-based gene prediction tool that uses a convolutional neural network (CNN) to predict genes in eukaryotic genomes. Helixer is trained on a wide range of eukaryotic genomes and can predict genes in both plant and animal genomes. Helixer can predict genes wihtout any extrinisic information such as RNA-seq data or homology information, purely based on the sequence of the genome.
1. Installation
Section titled “1. Installation”Helixer is available as a Singularity container. You can pull the container using the following command:
singularity pull docker://gglyptodon/helixer-docker:helixer_v0.3.3_cuda_11.8.0-cudnn8This will create helixer_v0.3.3_cuda_11.8.0-cudnn8.sif file in the current directory. We will rename this file to helixer.sif for simplicity.
2. Downloading trained models
Section titled “2. Downloading trained models”Helixer requires a trained model to predict genes. With the included script fetch_helixer_models.py you can download models for specific lineages. Currently, models are available for the following lineages:
land_plantvertebrateinvertibratefungi
There are instructions to train your own models as well as fine tune the existing models using the RNAseq data for the species of interest. But for this tutorial, we will just use the pre-trained models.
singularity exec helixer.sif fetch_helixer_models.py --allThis will download all lineage models in the models directory. You can also download models for specific lineages using the --lineage option.
The files downloaded will be in the following structure:
Directorymodels
Directoryfungi
- fungi_v0.3_a_0100.h5
Directoryinvertebrate
- invertebrate_v0.3_m_0100.h5
Directoryland_plant
- land_plant_v0.3_a_0080.h5
- model_list.csv
Directoryvertebrate
- vertebrate_v0.3_m_0080.h5
3. Running Helixer
Section titled “3. Running Helixer”Helixer requires GPU for prediction. For running Helixer, you need to request a GPU node. You will also need the genome sequence in fasta format. For this tutorial, we will use Maize genome (Zea mays subsp. mays), and use the land_plant model to predict genes.
Donwload the genome sequence:
wget https://download.maizegdb.org/Zm-B73-REFERENCE-NAM-5.0/Zm-B73-REFERENCE-NAM-5.0.fa.gzgunzip Zm-B73-REFERENCE-NAM-5.0.fa.gzRun Helixer:
genome=Zm-B73-REFERENCE-NAM-5.0.faspecies="Zea mays subsp. mays"output=B73-helixer.gffsingularity exec \ --bind ${RCAC_SCRATCH} \ --nv helixer.sif Helixer.py \ --lineage land_plant \ --fasta-path ${genome} \ --species ${species} \ --gff-output-path ${output}The GFF format output had 41,923 genes predicted using Helixer. You can view the various features in the gff file using the following command:
grep -v "^#" B73-helixer.gff | cut -f 3 | sort | uniq -coutputs:
2,19,488 CDS2,41,560 exon 52,607 five_prime_UTR 41,923 gene 41,923 mRNA 52,298 three_prime_UTR4. Comparing and benchmarking
Section titled “4. Comparing and benchmarking”We will download the reference annotations for Maize genome from MaizeGDB and compare the predicted genes with the reference annotations.
wget https://download.maizegdb.org/Zm-B73-REFERENCE-NAM-5.0/Zm-B73-REFERENCE-NAM-5.0.gff3.gzgunzip Zm-B73-REFERENCE-NAM-5.0.gff3.gzProcessing GFF output
The output generated by Helixer is in GFF format. However we might have to format it further to make it compatible with our processing workflow. Here are the steps to process the GFF output:
# sanitize the gff3 fileml purgeml biocontainersml genometoolsgff3_file=B73-helixer.gffgt gff3 \ -sort \ -tidy \ -setsource "helixer" \ -force -o B73-helixer_gt.gff3 \ ${gff3_file}
# standardizeml purgeml biocontainersml agatagat_convert_sp_gxf2gxf.pl \ -g B73-helixer_gt.gff3 \ -o B73-helixer_v1.0.gff3
# extract CDS and PEP sequnecesml purgeml biocontainersml cufflinksgffread B73-helixer_v1.0.gff3 \ -g Zm-B73-REFERENCE-NAM-5.0.fa \ -x B73-helixer_v1.0.cds.fasta \ -y B73-helixer_v1.0.pep.fastaA. BUSCO profiling
Section titled “A. BUSCO profiling”We will use BUSCO to profile the predicted genes and reference annotations.
ml biocontainersml buscobusco \ -i B73-helixer_v1.0.pep.fasta \ -c ${SLURM_CPUS_ON_NODE} \ -o B73-helixer_v1.0_busco \ -m prot \ -l poales_odb10 \ -fWe will also do this on the original reference annotations as well as the genome sequence.
Reference genome:
# reference genomebusco \ -i Zm-B73-REFERENCE-NAM-5.0.fa \ -c ${SLURM_CPUS_ON_NODE} \ -o Zm-B73-REFERENCE-NAM-5.0_busco \ -m genome \ -l poales_odb10 \ -fReference annotations:
# reference annotationswget https://download.maizegdb.org/Zm-B73-REFERENCE-NAM-5.0/Zm-B73-REFERENCE-NAM-5.0_Zm00001eb.1.protein.fa.gzgunzip Zm-B73-REFERENCE-NAM-5.0_Zm00001eb.1.protein.fa.gzbusco \ -i Zm-B73-REFERENCE-NAM-5.0_Zm00001eb.1.protein.fa \ -c ${SLURM_CPUS_ON_NODE} \ -o Zm-B73-REFERENCE-NAM-5.0_protein_busco \ -m prot \ -l poales_odb10 \ -fOnce the BUSCO profiling is done, the results can be compared.
Table 1: BUSCO results
| Category | Helixer | B73.v5 (MaizeGDB) | B73.v5 (Reference) |
|---|---|---|---|
| Complete BUSCOs | 4,808 (98.2%) | 4,729 (96.6%) | 4,807 (98.2%) |
| Single-Copy BUSCOs | 4,053 (82.8%) | 2,123 (43.4%) | 4,053 (82.8%) |
| Duplicated BUSCOs | 755 (15.4%) | 2606 (53.2%) | 754 (15.4%) |
| Fragmented BUSCOs | 34 (0.7%) | 39 (0.8%) | 10 (0.2%) |
| Missing BUSCOs | 54 (1.1%) | 128 (2.6%) | 79 (1.6%) |
| Total BUSCOs | 4,896 | 4,896 | 4,896 |
or visualized as a stacked bar plot:
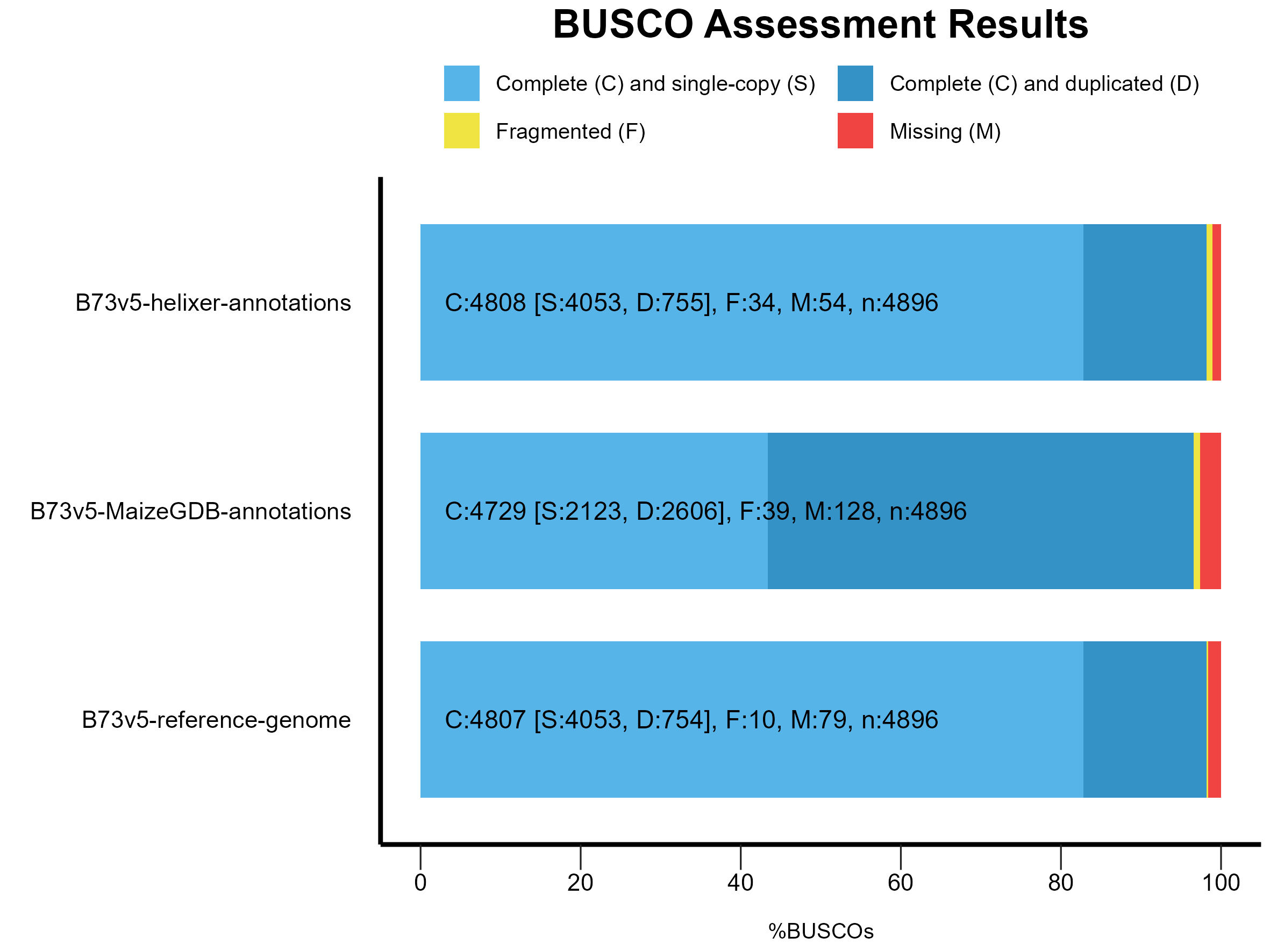
Figure 1: BUSCO results for Helixer, B73.v5 (MaizeGDB) and B73.v5 (Reference) annotations.
B. Comparing annotations
Section titled “B. Comparing annotations”We will use mikado to compare the annotations. Mikado is a tool to compare and merge gene annotations. We will use the mikado compare command to compare the annotations. We will filter V5 annotations to only include primary transcripts and only compare protein coding genes.
conda activate mikadomikado compare \ --protein-coding \ -r B73.v5_primary-only.gff3 \ -p B73-helixer_v1.0.gff3 \ -o ref_NAM.B73v5-vs-prediction_HELIXERv1_compared \ --log compare.logThe output will be in the ref_NAM.B73v5-vs-prediction_HELIXERv1_compared directory. The summary.tsv file will have the comparison results.
Table 2: Mikado comparison results
| Comparison Level | Sensitivity (Sn) | Precision (Pr) | F1 Score |
|---|---|---|---|
| Base level | 81.90% | 75.29% | 78.46% |
| Exon level (stringent) | 53.21% | 42.82% | 47.46% |
| Exon level (lenient) | 75.93% | 61.02% | 67.66% |
| Splice site level | 85.06% | 65.89% | 74.26% |
| Intron level | 78.45% | 60.77% | 68.49% |
| Intron chain level (stringent) | 38.10% | 32.85% | 35.28% |
| Transcript level (>=80% base F1) | 36.89% | 34.98% | 35.91% |
| Gene level (>=80% base F1) | 36.89% | 34.98% | 35.91% |
Table 3: Summary of comparison
| Feature | Total Count | Missed Count (%) | Novel Count (%) |
|---|---|---|---|
| Transcripts | 39,756 | 7,539 (18.96%) | 8,306 (19.81%) |
| Genes | 39,756 | 7,539 (18.96%) | 8,306 (19.81%) |
C. Feature assignment
Section titled “C. Feature assignment”B73 has extensive expression data and this can be used to evaluate the gene predictions. If the annotations are accurate, more RNA-seq reads will align within the predicted features, making a higher proportion of assigned reads an indicator of annotation accuracy. Similarly, higher proportions of unassigned reads indicate potential inaccuracies in the annotations, suggesting that important features may be missing or incorrectly predicted.
Expression data for B73 was downloaded from MaizeGDB (as BAM files) and feature assignment was done using featureCounts from the subread package (both NAM.v5 and Helixer predictions).
ml biocontainersml subreadfeatureCounts \ -T 8 \ -a Zm-B73-REFERENCE-NAM-5.0_Zm00001eb.1.gff3 \ -o v5.str2 \ -s 2 \ -p --countReadPairs \ -t gene -g ID \ --tmpDir /dev/shm \ *.bam &> v5_gene-str2_PE.stdout
featureCounts \ -T 8 \ -a B73-helixer_v1.0.gff3 \ -o helixer.str2 \ -s 2 \ -p --countReadPairs \ -t gene -g ID \ --tmpDir /dev/shm \ *.bam &> helixer_gene-str2_PE.stdout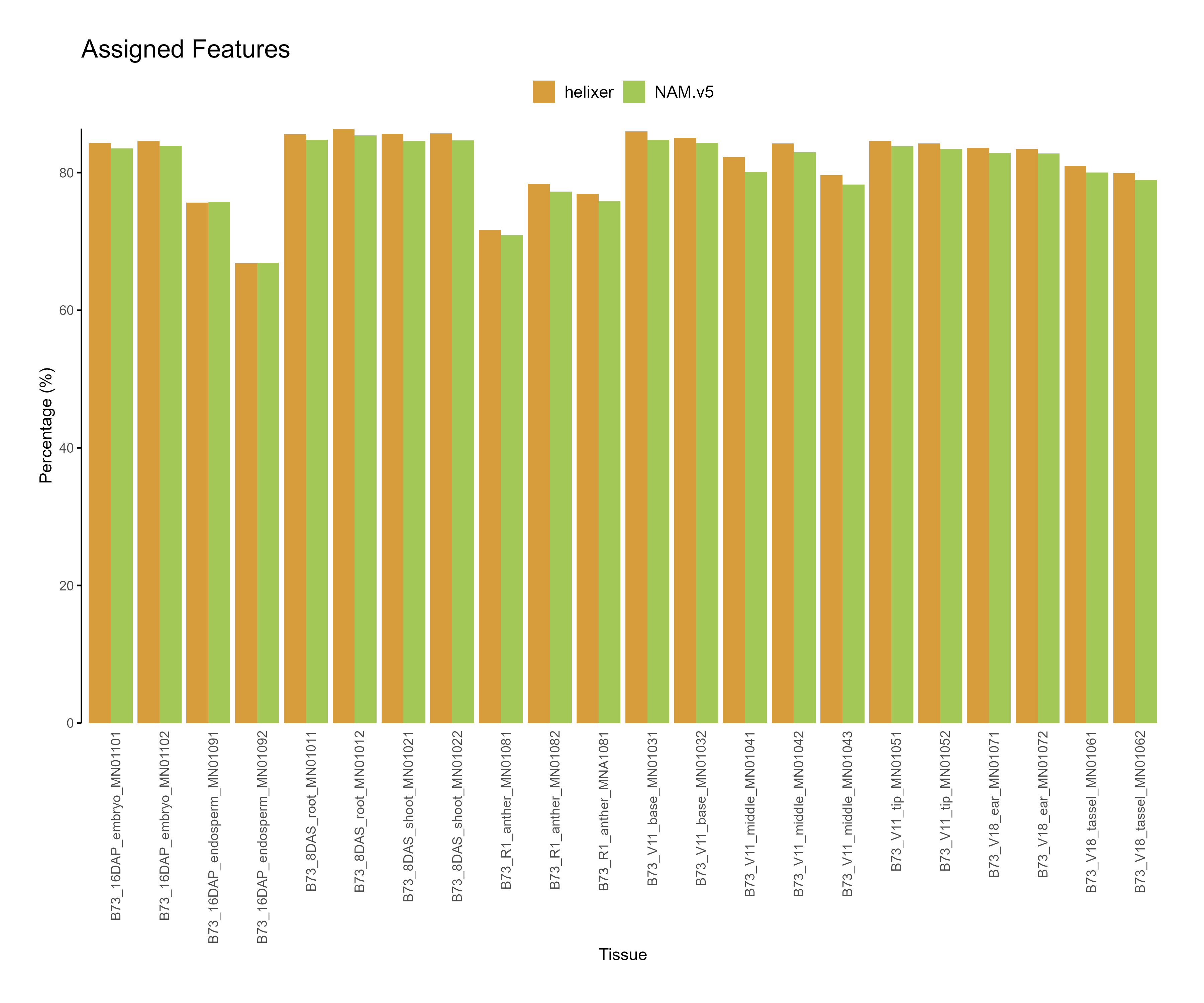
Figure 2: Feature assignment for B73.v5 and Helixer predictions. The reads assigned to helixer features are higher across tissues as compared to NAM.v5 annotations.
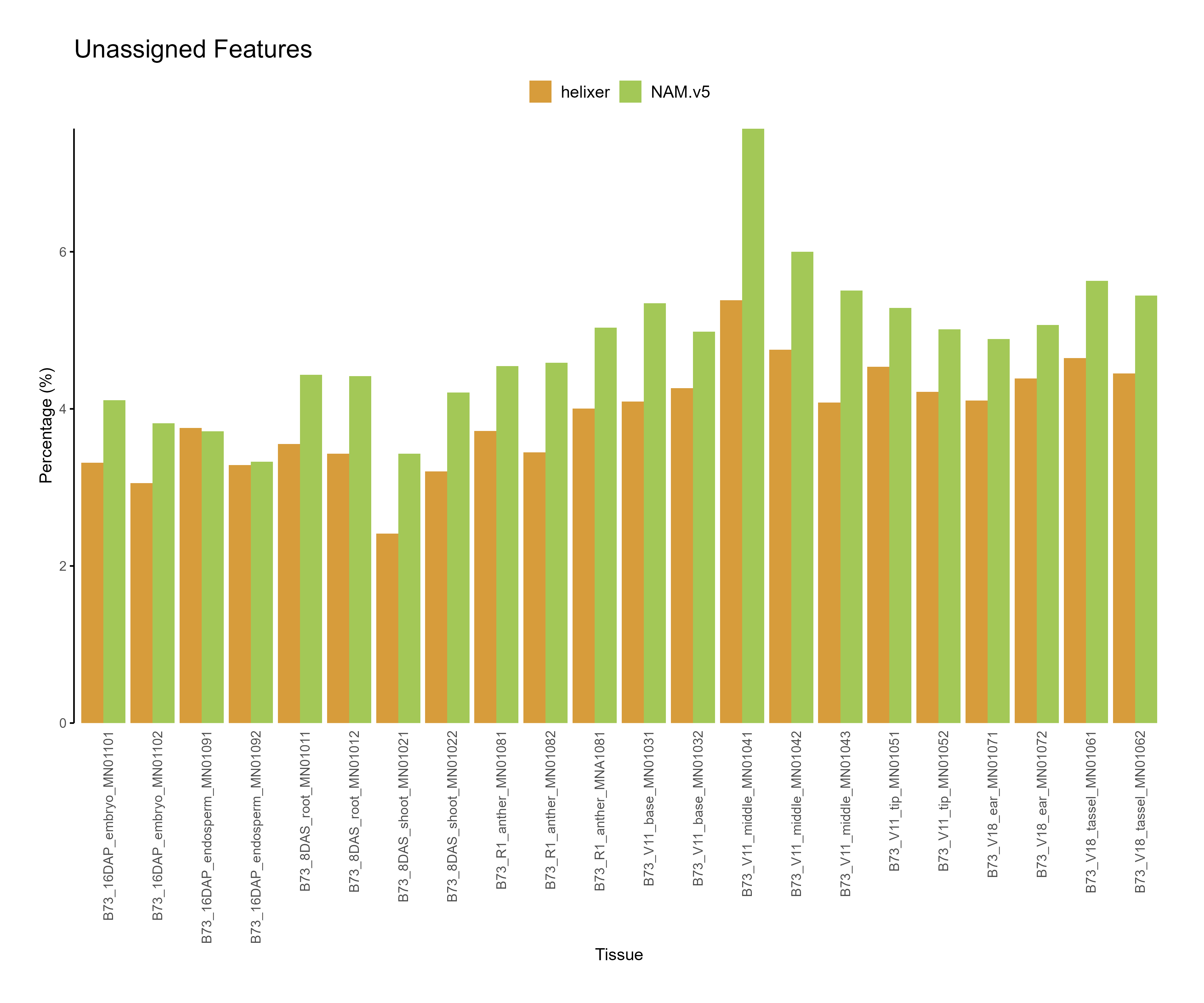
Figure 3: Feature assignment for B73.v5 and Helixer predictions. The unassigned reads are higher for NAM.v5 annotations across tissues.
The code used for parsing featureCounts summary files and generating the plots can be found here: parse_subreads.R
D. Functional annotation
Section titled “D. Functional annotation”The Eukaryotic Non-Model Transcriptome Annotation Pipeline (EnTAP) can be used for functional annotation of the predicted genes. We will use the entap pipeline determine proportion of genes with functional annotations in each predictions.
ml purgeml anacondaconda activate entapfor cds in B73-helixer_v1.0.cds.fasta Zm-B73-REFERENCE-NAM-5.0_Zm00001eb.1.cds.fa; doEnTAP \ --runP \ -i ${cds} \ -d ${RCAC_SCRATCH}/entap_db/bin/ncbi_refseq_plants.dmnd \ -d ${RCAC_SCRATCH}/entap_db/bin/uniprot_sprot.dmnd \ -t ${SLURM_CPUS_ON_NODE} \ --ini ${RCAC_SCRATCH}/entap_db/entap_config.inidone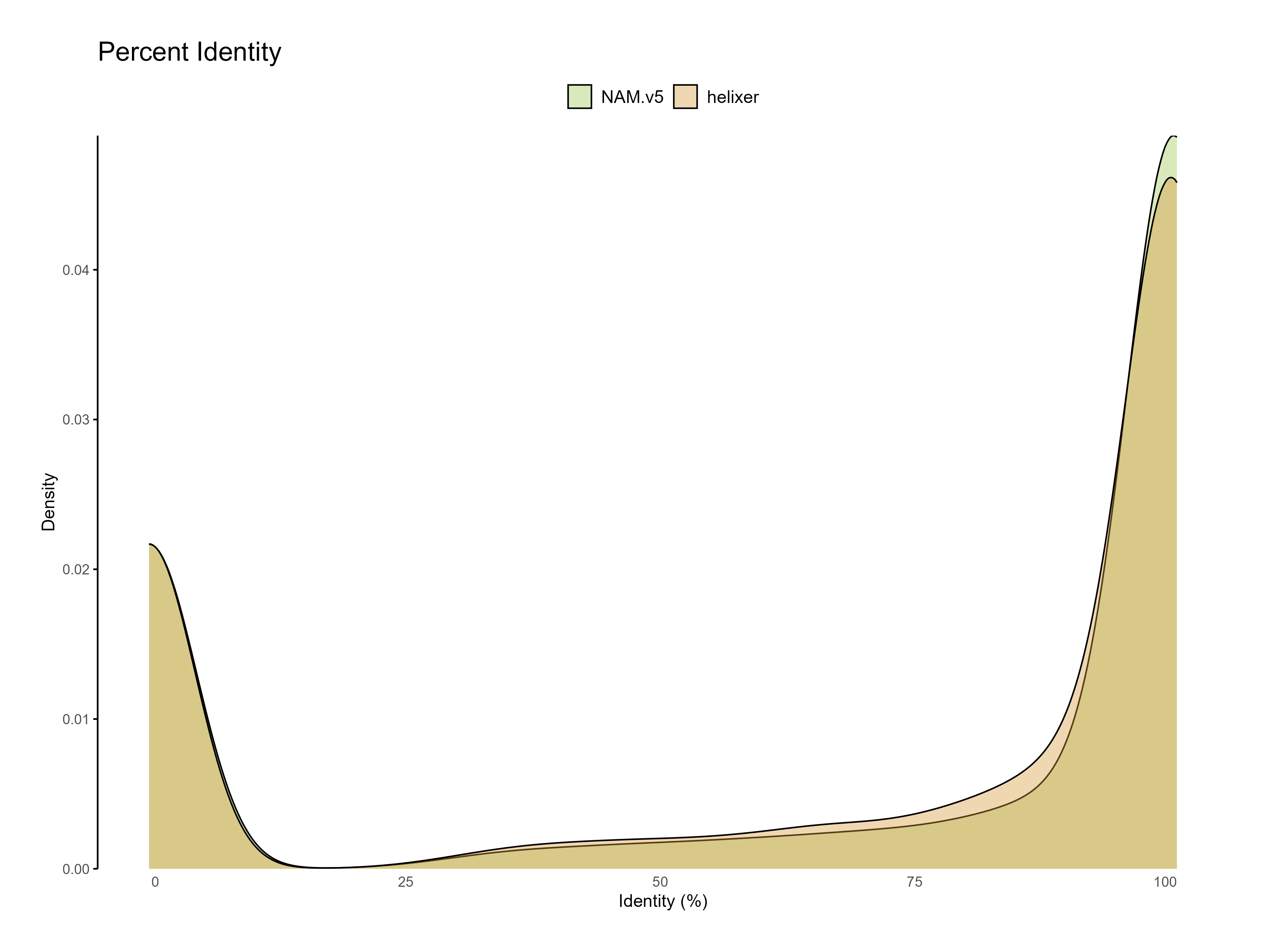
Figure 4: Percent identity of each predicted gene to the reference databases sequences (one hit per query).
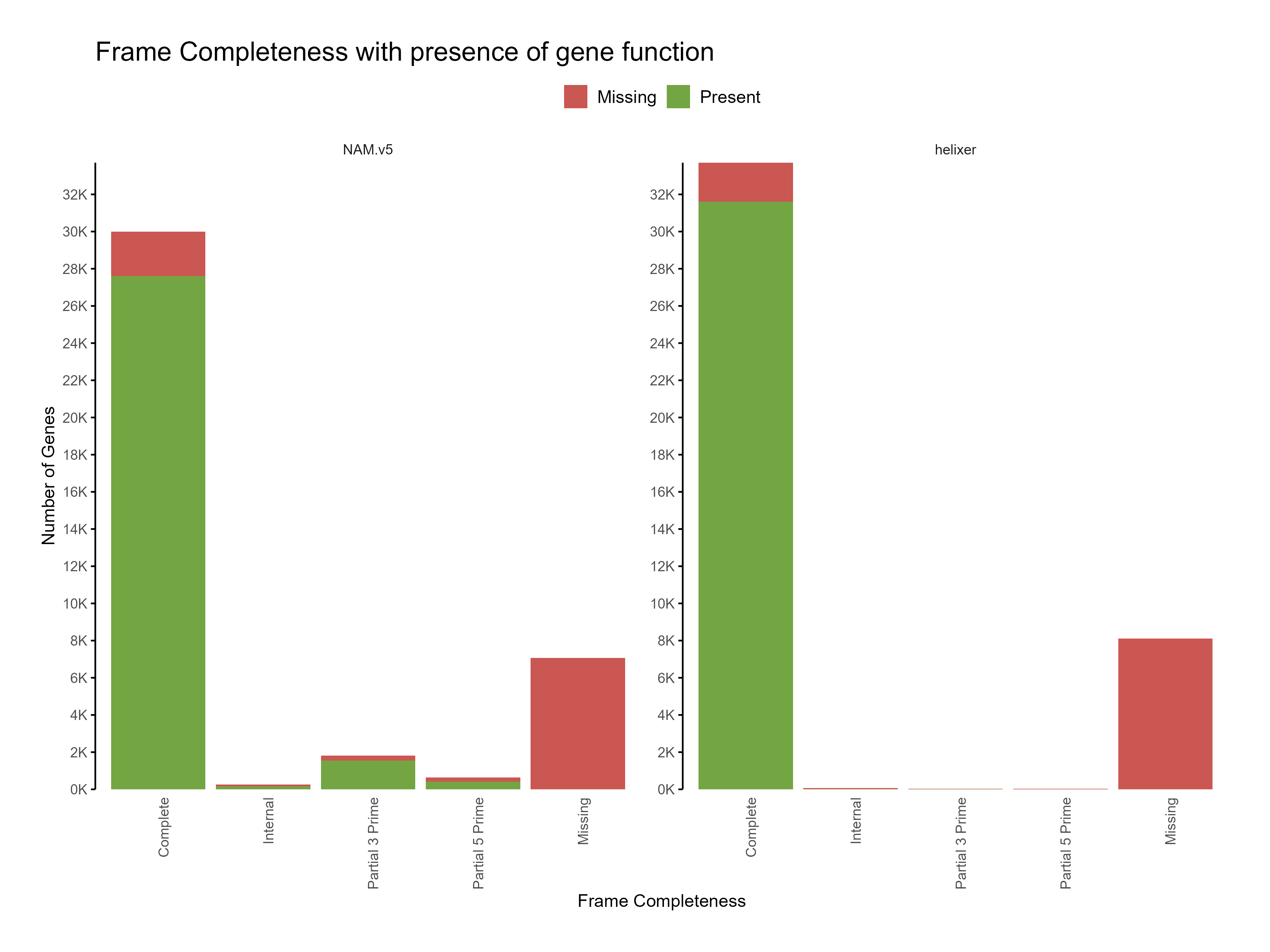
Figure 5: Distribution of frame completeness with presence of gene function across predictions
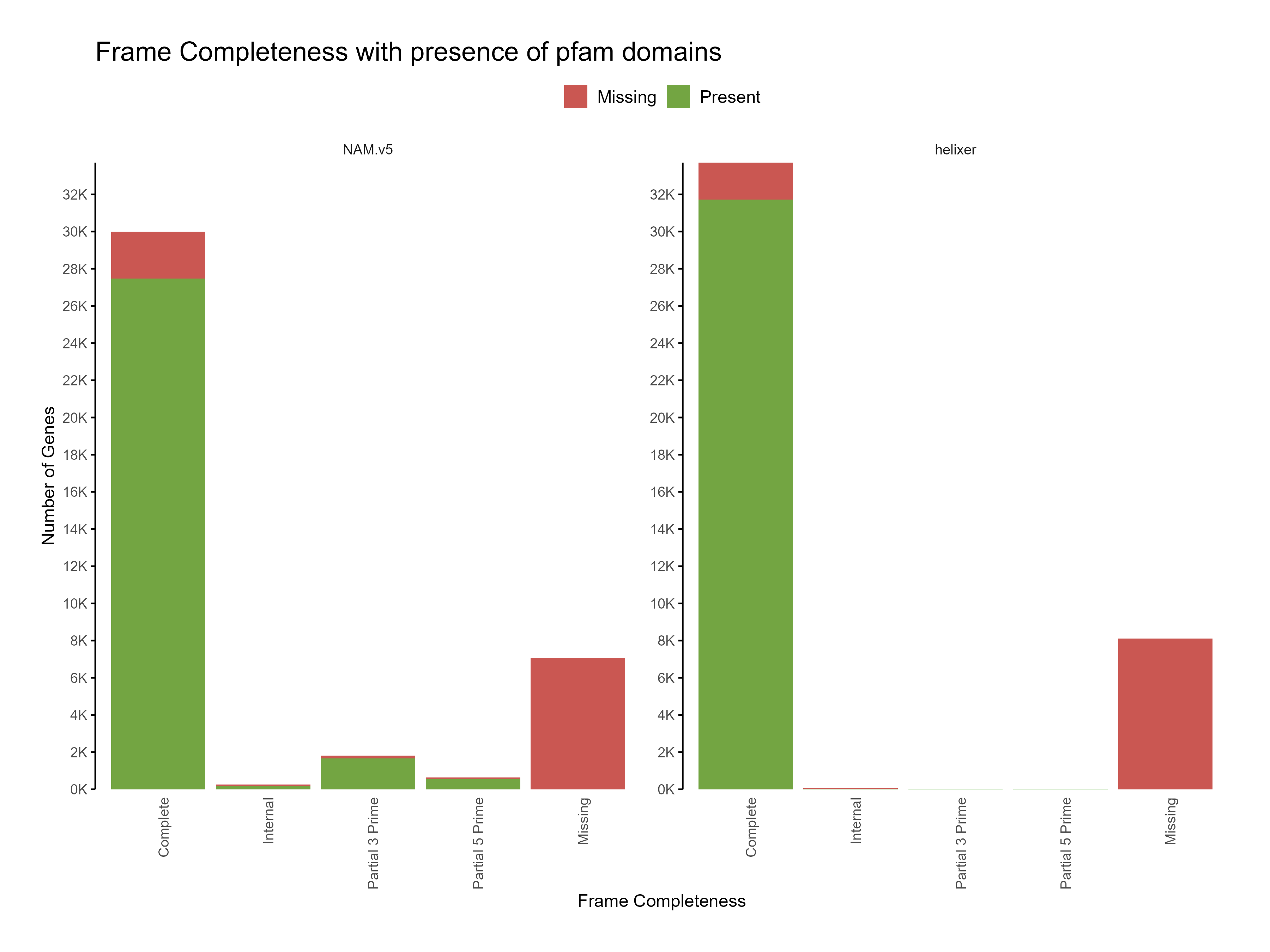
Figure 5: Distribution of frame completeness with presence of pfam domains across predictions
The code used for parsing EnTAP’s entap_results.tsv files and generating the plots can be found here: parse_subreads.R
E. Phylostrata analysis
Section titled “E. Phylostrata analysis”Phylostrata analysis can be used to determine the evolutionary age of the predicted genes. We will use the phylostrata package to determine the phylostrata of the predicted genes.
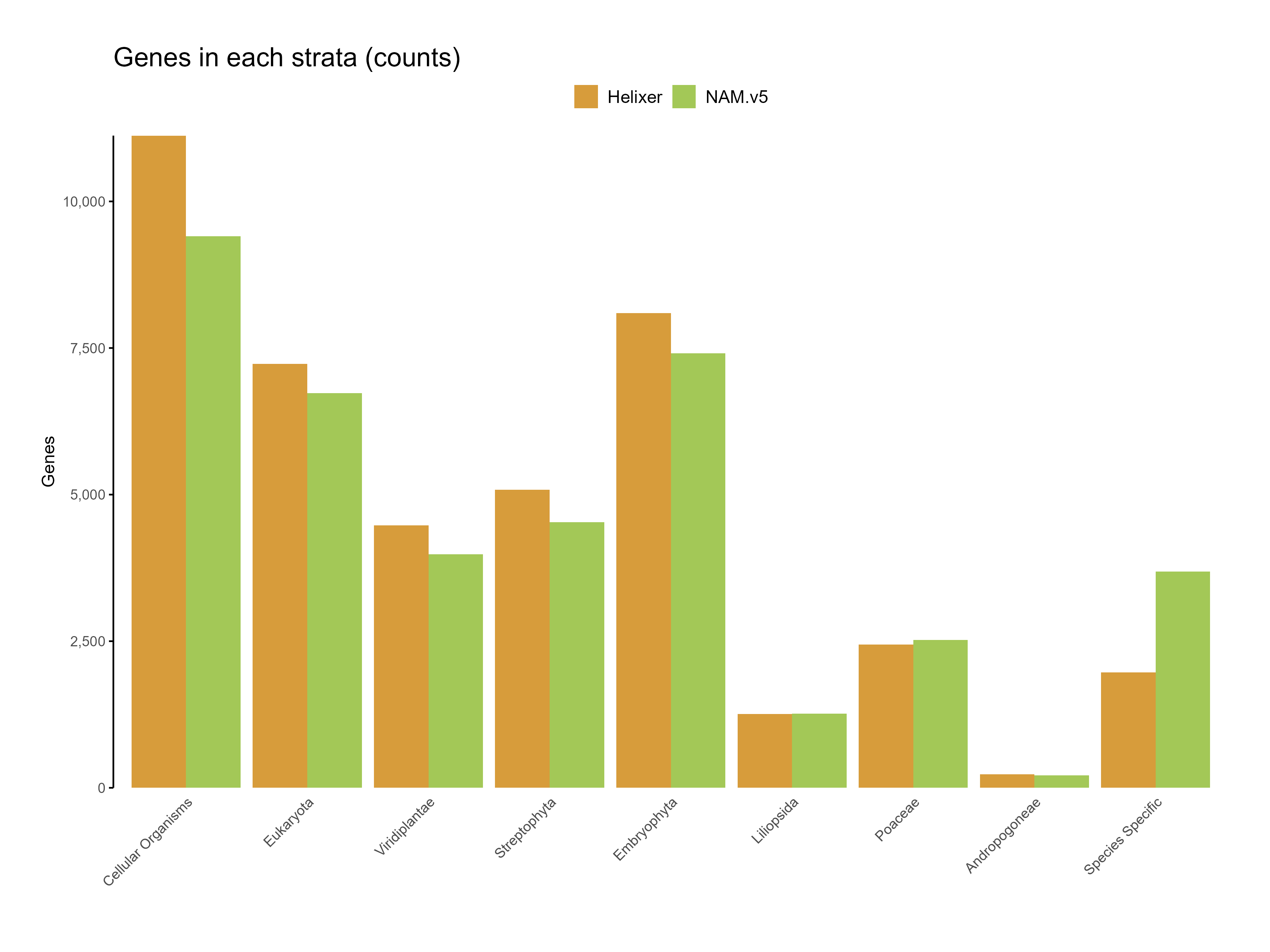
Figure 6: Genes in each starta for the predictions (count)
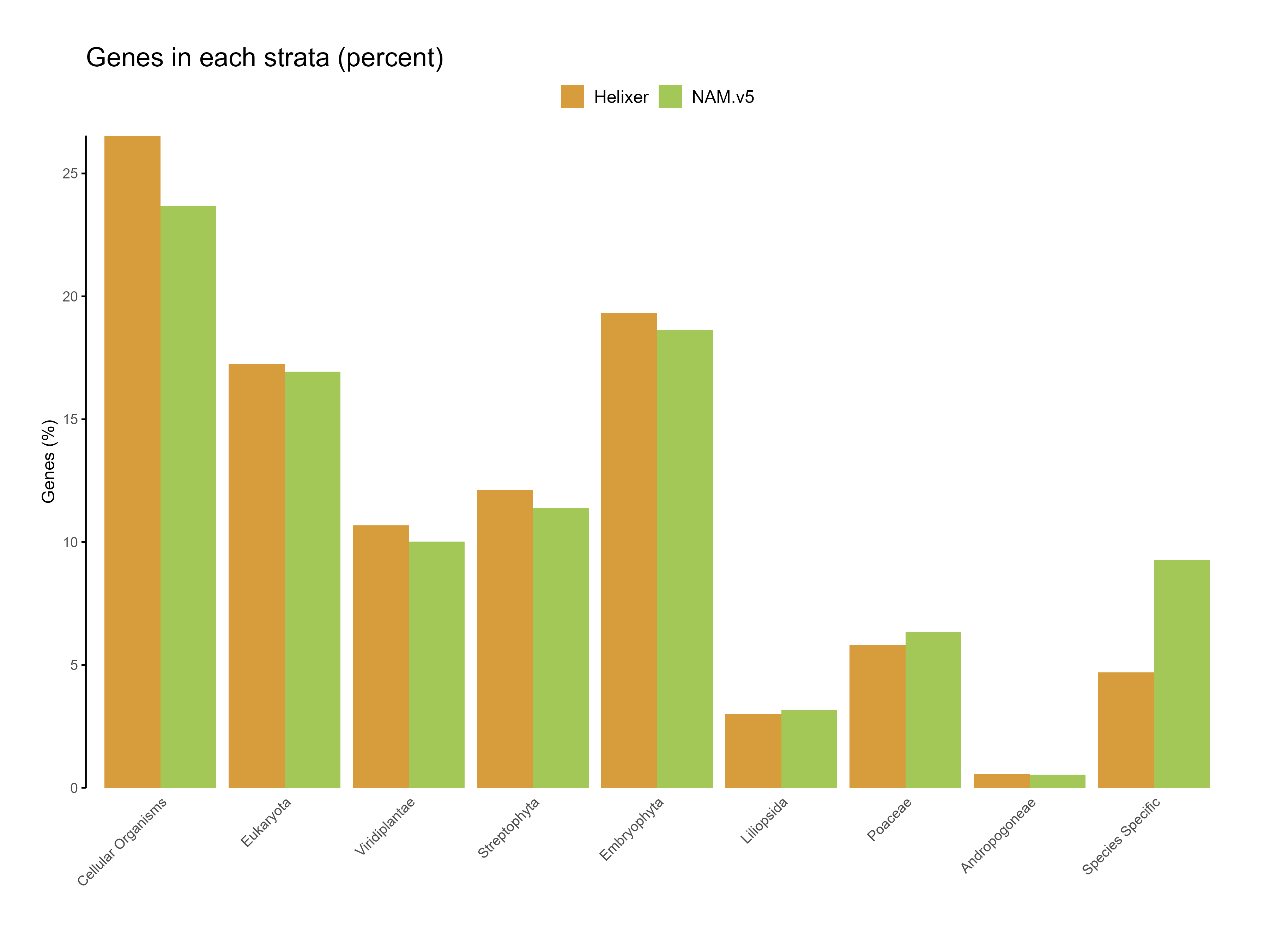
Figure 7: Genes in each starta for the predictions (percent)
The code used for parsing phylostrata analyses (phylostrata_run.R) and for plotting results (phylostrata_plot.R) are provided in the repository.
F. GFF3 stats comparison
Section titled “F. GFF3 stats comparison”You can generate comprehensive statistics for the GFF3 files using the agat_sp_statistics.pl script from the agat package.
ml biocontainersml agatagat_sp_statistics.pl \ -g B73-helixer_v1.0.gff3 \ -o B73-helixer_v1.0.statsagat_sp_statistics.pl \ -g Zm-B73-REFERENCE-NAM-5.0.gff3 \ -o Zm-B73-REFERENCE-NAM-5.0.statsThe statistics can be compared to identify differences in the gene predictions.
G. OMArk proteome assesment
Section titled “G. OMArk proteome assesment”OMArk provides measures of proteome completeness, characterizes the consistency of all protein coding genes with regard to their homologs, and identifies the presence of contamination from other species
omark_sif="${RCAC_SCRATCH}/omark/omark_0.3.0--pyh7cba7a3_0.sif"peptide="${RCAC_SCRATCH}/omark/Zm-B73-REFERENCE-NAM-5.0_Zm00001eb.1.protein.fa"database="${RCAC_SCRATCH}/omark/LUCA.h5"apptainer exec ${omark_sif} omamer search \ --db ${database} \ --query ${peptide} \ --out ${peptide%.*}.omamer \ --nthreads ${SLURM_CPUS_ON_NODE}# only needed for NAM.v5grep ">" ${peptide} |\ sed 's/>//g' |\ awk -F_ '{a[$1] = a[$1] ? a[$1]","$0 : $0} END {for (i in a) print a[i]}' > ${peptide%.*}.spliceapptainer exec ${omark_sif} omark \ --file ${peptide%.*}.omamer \ --database ${database} \ --outputFolder ${peptide%.*} \ --og_fasta ${peptide} \ --isoform_file ${peptide%.*}.spliceomark_sif="${RCAC_SCRATCH}/omark/omark_0.3.0--pyh7cba7a3_0.sif"peptide="${RCAC_SCRATCH}/omark/helixer_pep.fa"database="${RCAC_SCRATCH}/omark/LUCA.h5"apptainer exec ${omark_sif} omamer search \ --db ${database} \ --query ${peptide} \ --out ${peptide%.*}.omamer \ --nthreads ${SLURM_CPUS_ON_NODE}apptainer exec ${omark_sif} omark \ --file ${peptide%.*}.omamer \ --database ${database} \ --outputFolder ${peptide%.*} \ --og_fasta ${peptide} \ --isoform_file ${peptide%.*}.spliceTable 4: Conserved HOGs analysis for completeness assessment of gene sets in Helixer and NAM.v5 This table provides a completeness assessment of the gene sets for both Helixer and NAM.v5 based on conserved Hierarchical Orthologous Groups (HOGs) within the Panicoideae clade. The categories include the number of single-copy genes, duplicated genes, and missing genes, with a distinction between expected and unexpected duplications. The assessment serves as a proxy for evaluating the gene repertoire completeness and duplication events in each dataset.
| Category | Helixer Count | Helixer (%) | NAM.v5 Count | NAM.v5 (%) |
|---|---|---|---|---|
| Single | 13,072 | 63.75% | 9,150 | 44.62% |
| Duplicated | 5,874 | 28.65% | 2,240 | 10.92% |
| Duplicated, Unexpected | 2,150 | 10.49% | 990 | 4.83% |
| Duplicated, Expected | 3,724 | 18.16% | 1,250 | 6.10% |
| Missing | 1,559 | 7.60% | 9,115 | 44.45% |
| Total HOGs | 20,505 | 100.00% | 20,505 | 100.00% |
Table 5: Consistency assessment of annotated protein-coding genes in Helixer and NAM.v5 proteomes. This table presents the consistency assessment of annotated protein-coding genes within the Helixer and NAM.v5 proteomes. The categories evaluate gene placements as “Consistent” with the ancestral lineage, “Inconsistent” (potential misannotations), “Contaminants” (genes from other lineages), and “Unknown” (unmatched genes). Subcategories include partial hits and fragmented proteins, which may indicate issues with gene models, annotation, or sequence integrity.
| Category | Helixer Count | Helixer (%) | NAM.v5 Count | NAM.v5 (%) |
|---|---|---|---|---|
| Total Proteins | 41,923 | 100.00% | 39,756 | 100.00% |
| Consistent | 38,997 | 93.02% | 20,208 | 50.83% |
| Consistent, partial hits | 5,412 | 12.91% | 3,541 | 8.91% |
| Consistent, fragmented | 3,094 | 7.38% | 2,627 | 6.61% |
| Inconsistent | 757 | 1.81% | 1,910 | 4.80% |
| Inconsistent, partial hits | 291 | 0.69% | 335 | 0.84% |
| Inconsistent, fragmented | 279 | 0.67% | 921 | 2.32% |
| Contaminants | 0 | 0.00% | 0 | 0.00% |
| Contaminants, partial hits | 0 | 0.00% | 0 | 0.00% |
| Contaminants, fragmented | 0 | 0.00% | 0 | 0.00% |
| Unknown | 2,169 | 5.17% | 17,638 | 44.37% |
H. CDS assesments (GC, length, start/stop)
Section titled “H. CDS assesments (GC, length, start/stop)”We can use bioawk get relavant stats on the CDS sequences predicted by Helixer and compare them with the reference annotations.
for f in *_cds.fa; do bioawk -c fastx 'BEGIN{OFS="\t"}{ print $name,length($seq),gc($seq),substr($seq,0,3),reverse(substr(reverse($seq),0,3)) }' $f > ${f%.*}.info;doneThe plots were generated using the following R script: cds_assesment.R
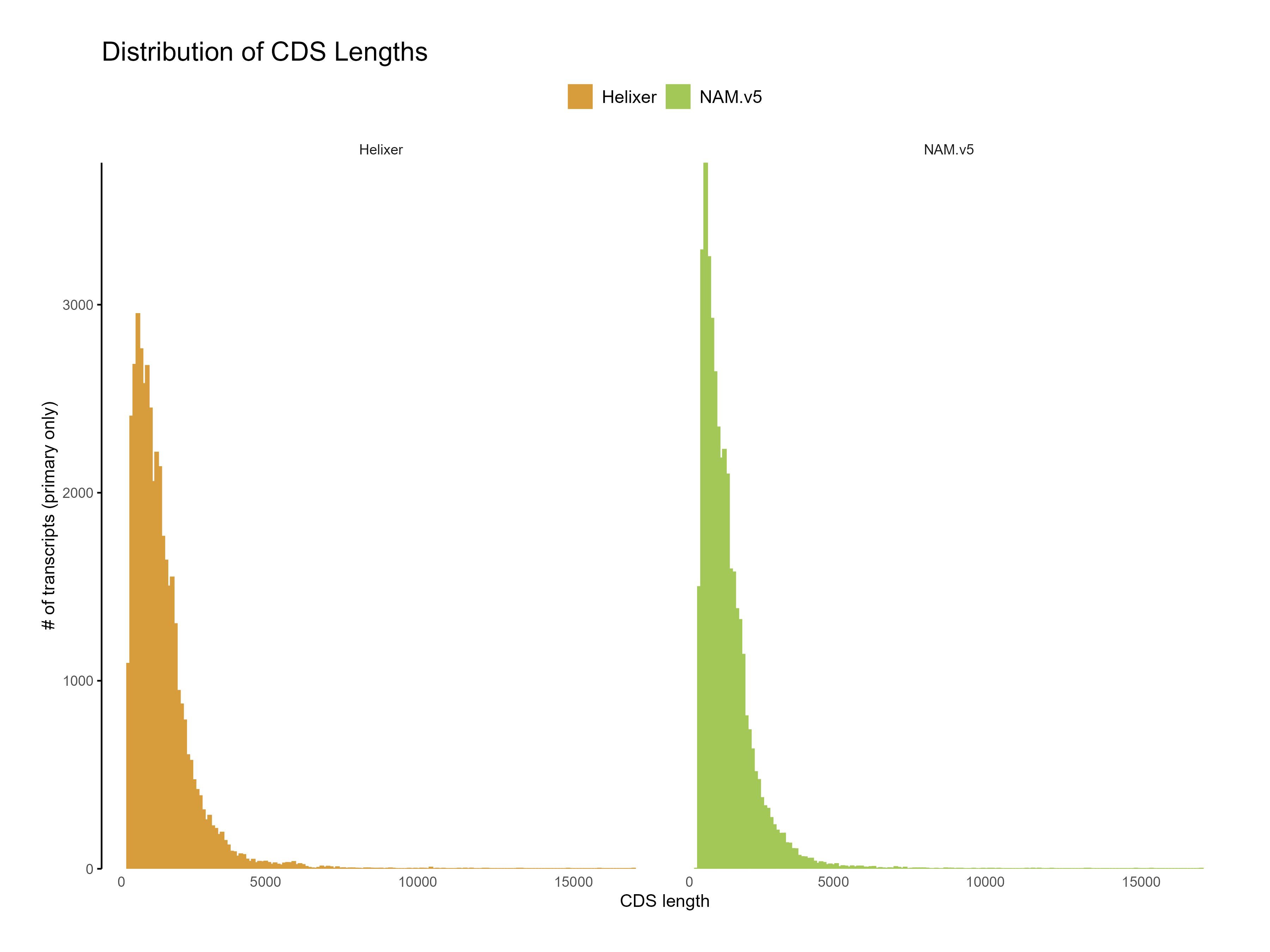
Figure 8: Distribution of CDS length for Helixer and NAM.v5 predictions. The Helixer predictions have a lower proportion of shorter CDS lengths compared to NAM.v5 annotations.
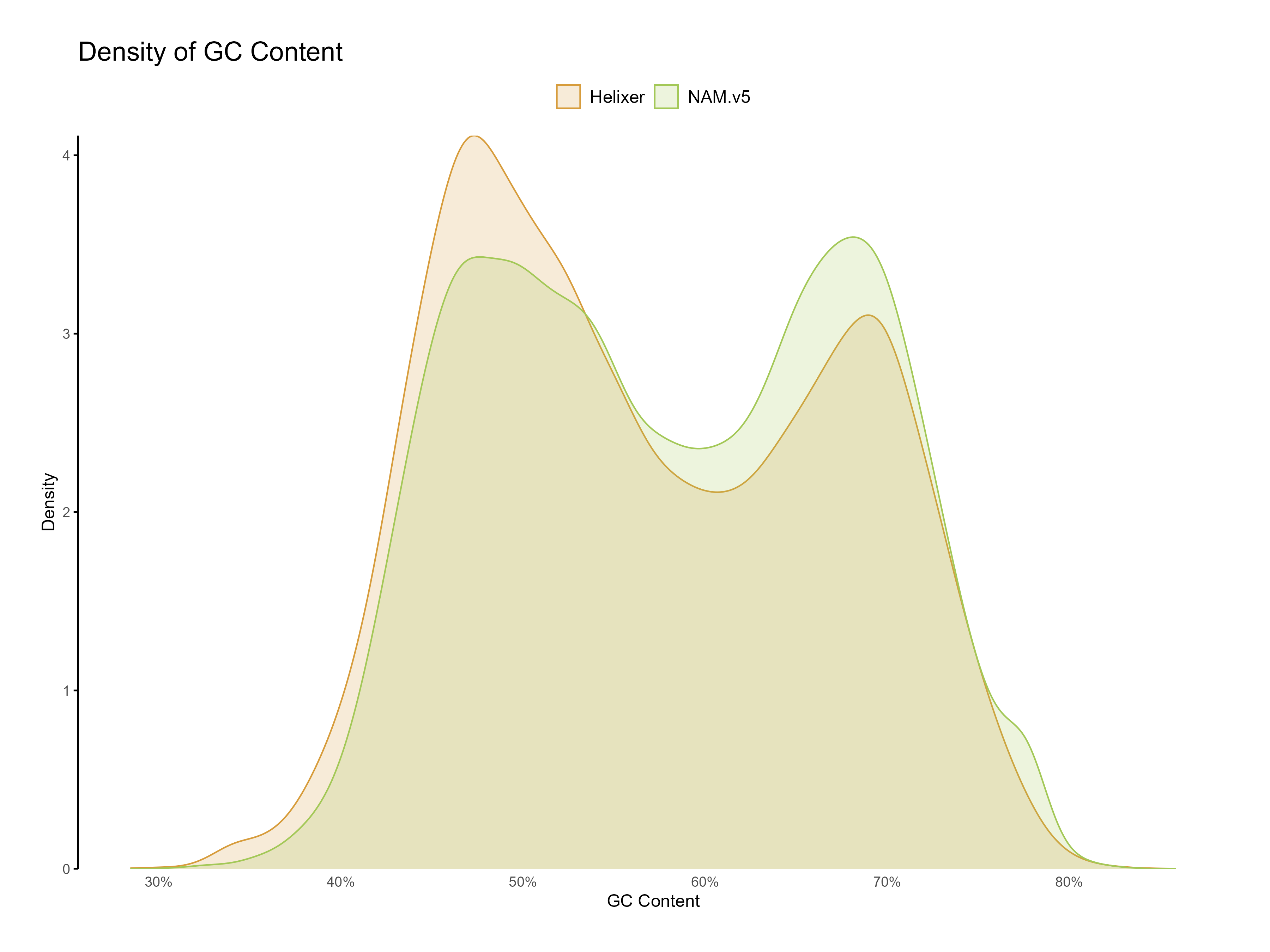
Figure 9: Distribution of GC content for Helixer and NAM.v5 predictions. Both predictions have a classic dual GC peak typical to grasses.
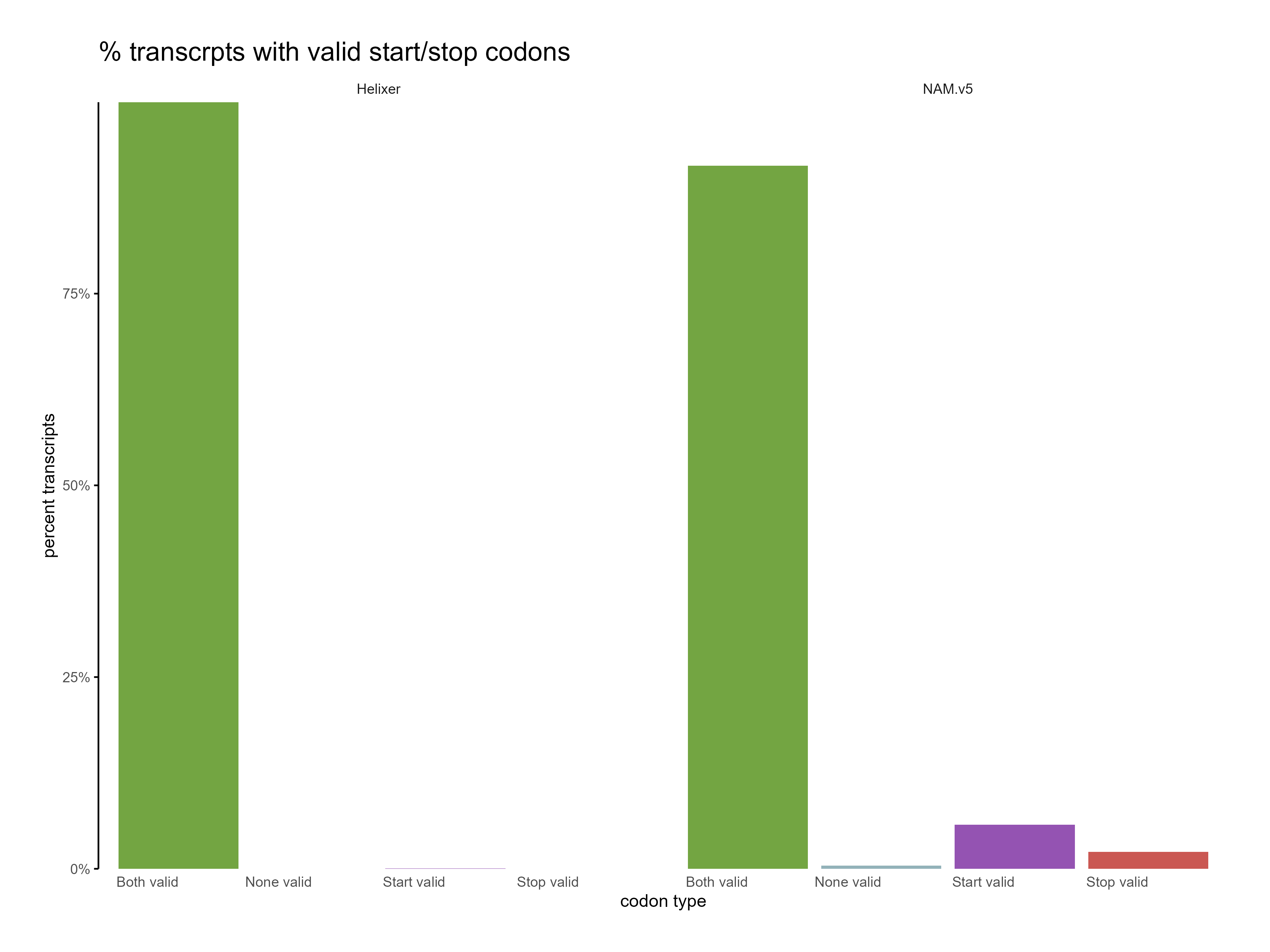
Figure 10: Distribution of start and stop codons for Helixer and NAM.v5 predictions. The Helixer predictions have a higher proportion of valid start and stop codons.
Key Points
Section titled “Key Points”-
Helixer’s strength in conserved genes: Helixer effectively predicts genes in deeply conserved phylostrata, indicating strong performance in identifying core, evolutionarily stable genes across eukaryotic lineages.
-
Limited novel gene detection: Helixer shows lower accuracy in detecting species-specific genes, suggesting that identifying novel or rapidly evolving genes may not be its primary strength.
-
Pre-trained models for versatile Use: With pre-trained models for various lineages (plants, animals, fungi), Helixer is accessible for multiple research applications without requiring additional RNA-seq or homology data.
-
Simplicity of Gene Prediction: Helixer allows for rapid and straightforward gene prediction with a single command, requiring no external data or repeat masking. It can predict genes in complex, highly repetitive genomes like maize in under four hours. This simplicity, combined with easy installation, has the potential to revolutionize gene prediction workflows.
-
Ideal for Comparative Genomics: Helixer’s robust detection of conserved genes makes it a valuable tool for studies focused on comparative genomics and understanding fundamental gene functions.
-
Potential for Improvement: The presence of unassigned reads across samples and limited species-specific gene detection highlight areas for future model refinement, possibly with custom training for novel species.
5. References
Section titled “5. References”-
Holst F, Bolger A, Günther C, Maß J, Kindel F, Triesch S, Kiel N, Saadat N, Ebenhöh O, Usadel B,Schwacke R, Bolger M, Weber APM, Denton AK Helixer - de novo prediction of primary eukaryotic gene models combining deep learning and aHidden Markov model. bioRxiv. 2023 Feb 06: 2023.02.06.527280. Preprint. (not been certified by peer review)
-
Stiehler F, Steinborn M, Scholz S, Dey D, Weber APM, Denton AK Helixer: cross-species gene annotation of large eukaryotic genomes using deep learning. Bioinformatics. 2020 Dec, 36(22-23): 5291-5298.