Downloading and organizing files
Last updated on 2026-08-04 | Edit this page
Estimated time: 35 minutes
Overview
Questions
- What files are required to process raw RNA seq reads?
- Where do we obtain raw reads, reference genomes, annotations, and transcript sequences?
- How should project directories be organized to support a smooth workflow?
- What practical considerations matter when downloading and preparing RNA seq data?
Objectives
- Identify the essential inputs for RNA seq analysis: FASTQ files, reference genome, annotation, and transcript sequences.
- Learn where and how to download RNA seq datasets and reference materials.
- Create a clean project directory structure suitable for quality control, alignment, and quantification.
- Understand practical issues such as compressed FASTQ files and adapter trimming.
Introduction
Before we can perform quality control, mapping, or quantification, we must first gather the files required for RNA seq analysis and organize them in a reproducible way. This episode introduces the essential inputs of an RNA seq workflow and demonstrates how to obtain and structure them on a computing system.
We begin by discussing raw FASTQ reads, reference genomes, annotation files, and transcript sequences. We then download the dataset used throughout the workshop and set up the directory structure that supports downstream steps.
Most FASTQ files arrive compressed as .fastq.gz files.
Modern RNA seq tools accept compressed files directly, so uncompressing
them is usually unnecessary.
Should I uncompress FASTQ files?
In almost all RNA-seq workflows, you should keep FASTQ files
compressed. Tools such as FastQC, HISAT2, STAR, Salmon, and fastp can
read .gz files directly. Keeping files compressed saves
space and reduces input and output overhead.
Many library preparation protocols introduce adapter sequences at the ends of reads. For most alignment based analyses, explicit trimming is not required because aligners soft clip adapter sequences automatically.
Should I trim adapters for RNA seq analysis?
In most cases, no. Aligners such as HISAT2 and STAR soft clip adapter
sequences. Overly aggressive trimming can reduce mapping rates or
distort read length distributions. Trimming is needed only for specific
applications such as transcript assembly where uniform read lengths are
important.
More information: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4766705/
The workshop dataset: p53-mediated response to ionizing radiation
For this workshop, we use a subset of a publicly available dataset (GEO accession GSE71176) that investigates the transcriptional response to ionizing radiation (IR) in mouse B cells. The original study by Tonelli et al. (2015) examined how the tumor suppressor p53 (encoded by Trp53) regulates gene expression following DNA damage.
Biological context
The TP53 gene (called Trp53 in mice) encodes the p53 protein, often called the “guardian of the genome.” When cells experience DNA damage, such as double-strand breaks caused by ionizing radiation, p53 activates transcriptional programs that lead to:
- Cell cycle arrest (allowing time for DNA repair)
- Apoptosis (programmed cell death if damage is irreparable)
- Senescence (permanent growth arrest)
- Metabolic reprogramming
Mutations in TP53 are among the most common alterations in human cancers, occurring in approximately 50% of all tumors. Understanding p53-regulated genes is therefore central to cancer biology.
Experimental design
The full dataset includes wild-type (WT) and p53-knockout (KO) mouse B cells, with and without IR treatment (7 Gy, 4 hours post-exposure). For simplicity, we will focus on 8 samples from wild-type B cells only:
| Condition | Description | Samples | Replicates |
|---|---|---|---|
| WT_mock (control) | Wild-type B cells, no radiation | 4 | SRR2121778-81 |
| WT_IR (treatment) | Wild-type B cells, 4h post 7Gy IR | 4 | SRR2121786-89 |
This balanced design (n=4 per group) allows us to identify genes that respond to ionizing radiation in cells with functional p53.
Expected biological results
Because p53 is a transcription factor activated by DNA damage, we expect to see upregulation of canonical p53 target genes in IR-treated samples:
Genes expected to be upregulated (IR vs mock):
- Cdkn1a (p21) - cell cycle arrest
- Bax, Bbc3 (Puma), Pmaip1 (Noxa) - pro-apoptotic
- Mdm2 - negative feedback regulator of p53
- Gadd45a - DNA damage response
- Fas, Tnfrsf10b (DR5) - death receptor signaling
Pathways expected to be enriched:
- p53 signaling pathway
- Apoptosis
- Cell cycle
- DNA damage response
The presence of these expected results serves as a positive control that our analysis pipeline is working correctly.
Why this dataset?
This dataset is ideal for teaching because:
- Simple two-group comparison: Control vs. treatment with balanced replicates
- Clear biological interpretation: DNA damage response is well-characterized
- Expected positive controls: Known p53 targets should appear as top hits
- Clinical relevance: p53 biology is fundamental to cancer research
- Model organism: Mouse with excellent genome annotation
Project organization
Before downloading files, we create a reproducible directory layout for raw data, scripts, mapping outputs, and count files.
Create the directories needed for this episode
Create a working directory for the workshop (e.g.,
rnaseq-workshop). Inside it, create four
subdirectories:
-
data: raw FASTQ files, reference genome, annotation -
scripts: custom scripts, SLURM job files -
results: alignment, counts and various other outputs
The resulting directory structure should look like this:
What files do I need for RNA-seq analysis?
You will need:
- Raw reads (FASTQ)
- Usually
.fastq.gzfiles containing nucleotide sequences and per-base quality scores.
- Usually
For alignment based workflows
- Reference genome (FASTA)
- Contains chromosome or contig sequences.
- Annotation file (GTF/GFF)
- Describes gene models: exons, transcripts, coordinates.
For transcript-level quantification workflows
- Transcript sequences (FASTA)
- Required for transcript-based quantification with tools such as Salmon, Kallisto, and RSEM.
Reference files must be consistent with each other (same genome version). To ensure a smooth analysis workflow, keep these files logically organized. Good directory structure minimizes mistakes, simplifies scripting, and supports reproducibility.
Downloading the data
We now download:
- the GRCm39 reference genome
- the corresponding GENCODE annotation
- transcript sequences (if using transcript based quantification)
- raw FASTQ files from SRA
Reference genome and annotation
We will use GENCODE GRCm39 (M38) as the reference.
Annotation
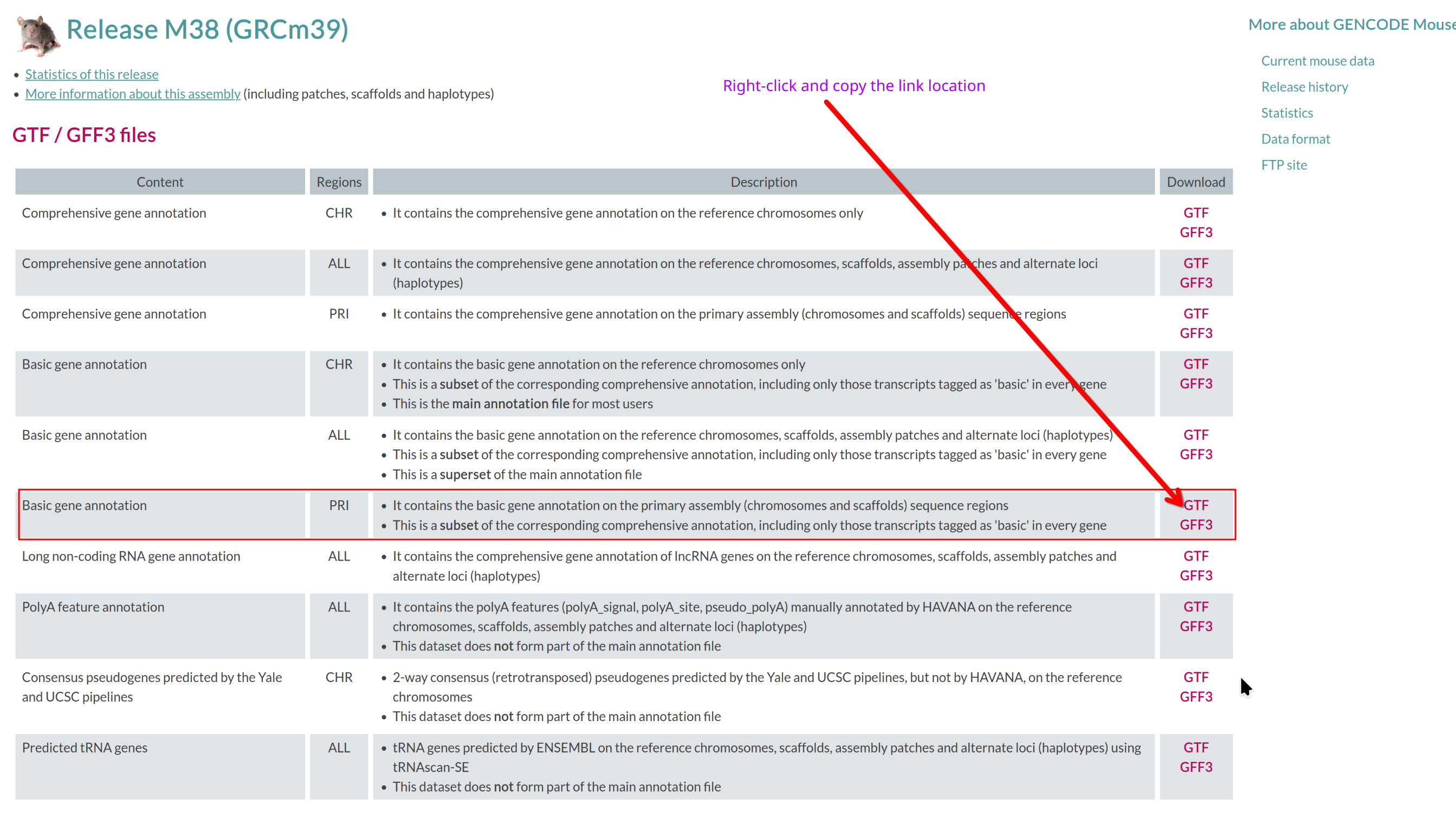
Visit the GENCODE mouse page: https://www.gencodegenes.org/mouse/.
Which annotation file (GTF/GFF3 files section) should you download, and
why?
Use the basic gene annotation in GTF format (e.g.,
gencode.vM38.primary_assembly.basic.annotation.gtf.gz).
It contains essential gene and transcript models without pseudogenes or
other biotypes not typically used for RNA-seq quantification.
Reference genome
Select the corresponding Genome sequence (GRCm39)
FASTA (e.g.,
GRCm39.primary_assembly.genome.fa.gz).

Transcript sequences
Transcript level quantification requires a transcript FASTA file
consistent with the annotation. GENCODE provides transcript sequences
for all transcripts in GRCm39
(gencode.vM38.transcripts.fa.gz).
Transcript sequence file
Where on the GENCODE page can you find the transcript FASTA file?
The transcript FASTA file is available in the same release directory
as the genome and annotation files (under FASTA files). It is named
gencode.vM38.transcripts.fa.gz.

Downloading all reference files
BASH
cd ${SCRATCH}/rnaseq-workshop
GTFlink="https://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_mouse/release_M38/gencode.vM38.primary_assembly.basic.annotation.gtf.gz"
GENOMElink="https://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_mouse/release_M38/GRCm39.primary_assembly.genome.fa.gz"
CDSLink="https://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_mouse/release_M38/gencode.vM38.transcripts.fa.gz"
# Download to 'data'
wget -P data ${GENOMElink}
wget -P data ${GTFlink}
wget -P data ${CDSLink}
# Uncompress
cd data
gunzip GRCm39.primary_assembly.genome.fa.gz
gunzip gencode.vM38.primary_assembly.basic.annotation.gtf.gz
gunzip gencode.vM38.transcripts.fa.gzThe transcript file header has multiple pieces of information which often interfere with downstream analysis. We create a simplified version containing only the transcript IDs.
Raw reads
-
Study: Genome-wide analysis of p53 transcriptional
programs in B cells upon exposure to genotoxic stress in
vivo
- Organism: Mus musculus (C57BL/6)
- Cell type: Primary B cells from spleen
- Design: Wild-type mock vs Wild-type IR-treated (n=4 per group)
- Treatment: 7 Gy ionizing radiation, harvested 4 hours post-exposure
- Sequencing: Illumina HiSeq 2000, paired-end 51bp
- GEO: GSE71176
- Reference: Tonelli et al. Oncotarget 2015 Sep 22;6(28):24611-26. PMID: 26372730
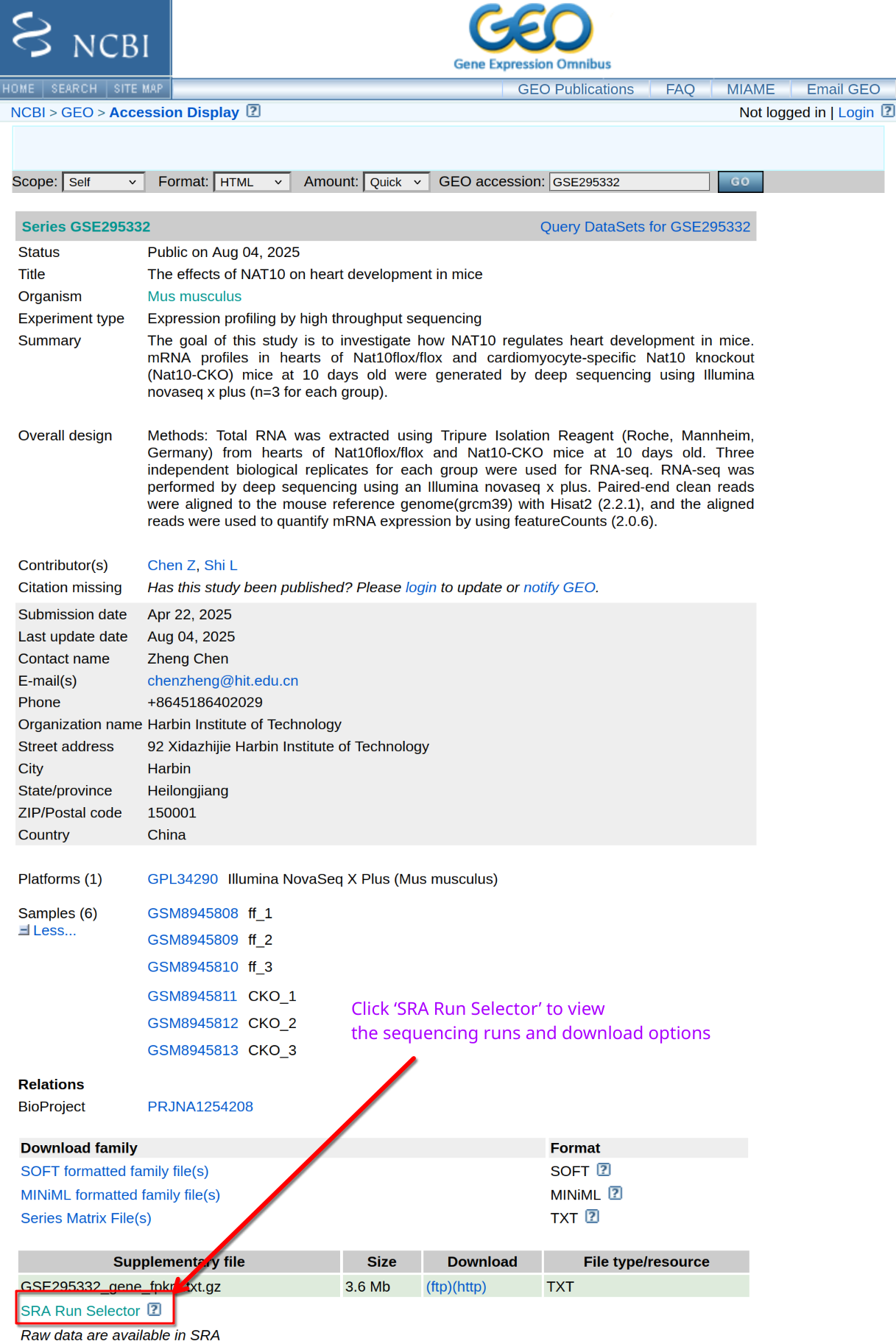
Where do you find FASTQ files?
GEO provides metadata and experimental details, but FASTQ files are stored in the SRA. How do you obtain the complete list of sequencing runs?
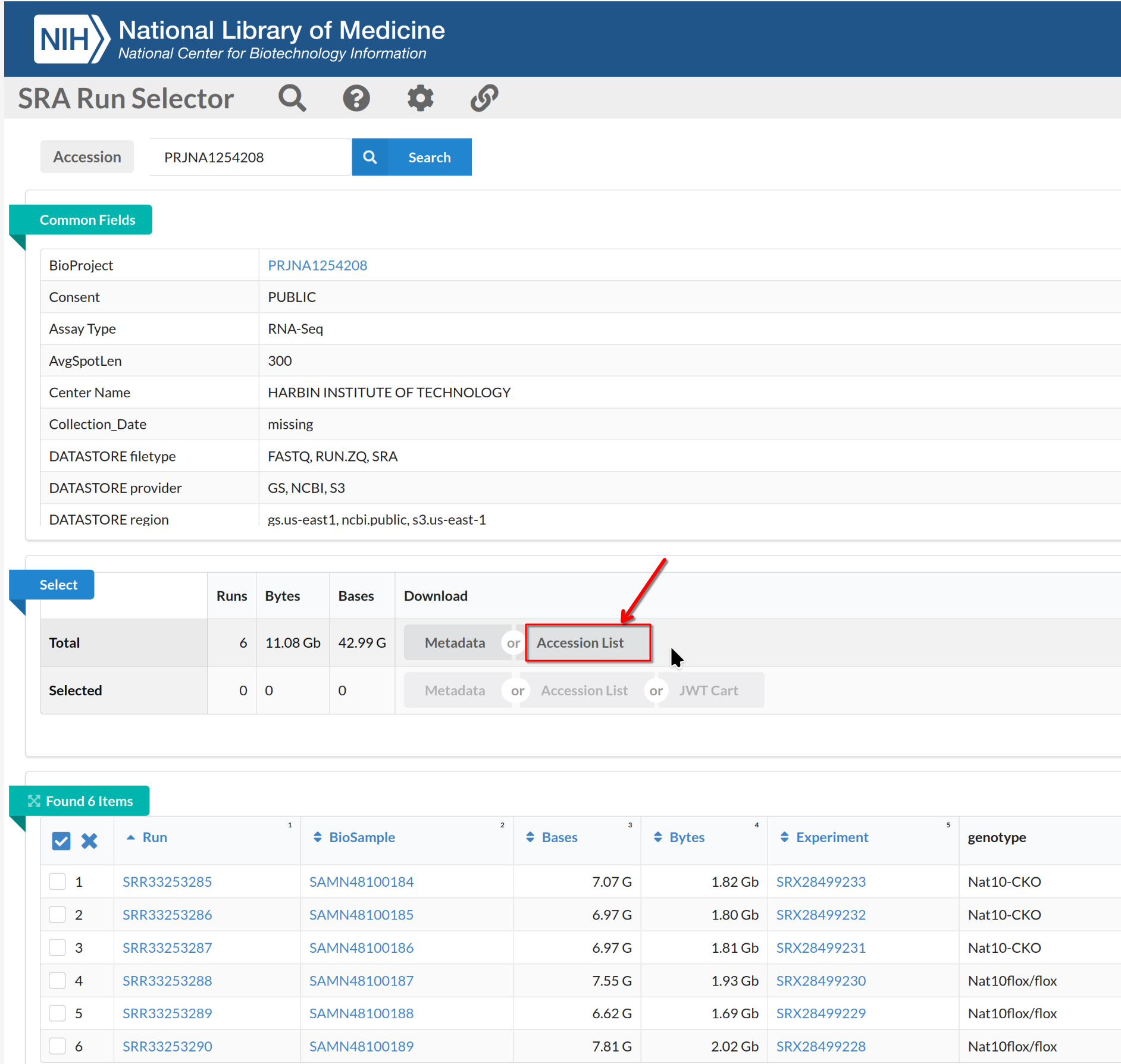
Navigate to the SRA page for the BioProject and use Run
Selector to list runs.
On the Run Selector page, select the accessions (SRR2121778-81,
SRR2121786-89), toggle the “selected” and click Accession
List to download the selected SRR accession IDs.
Download FASTQ files with SRA Toolkit
[DO NOT RUN THIS COMMAND DURING THE WORKSHOP - IT TAKES TOO LONG TO COMPLETE]. You already copied the pre-downloaded data to your scratch space.
BASH
sinteractive -A rcac-rnaseq -q standby -p cpu -N 1 -n 4 --time=1:00:00
cd ${SCRATCH}/rnaseq-workshop/data
module load biocontainers
module load sra-tools
while read SRR; do
fasterq-dump --threads 4 --progress $SRR;
gzip ${SRR}_1.fastq
gzip ${SRR}_2.fastq
done<SRR_Acc_List.txt
# rename files to meaningful names
# WT B cells - mock
mv SRR2121778_1.fastq.gz WT_Bcell_mock_rep1_R1.fastq.gz
mv SRR2121778_2.fastq.gz WT_Bcell_mock_rep1_R2.fastq.gz
mv SRR2121779_1.fastq.gz WT_Bcell_mock_rep2_R1.fastq.gz
mv SRR2121779_2.fastq.gz WT_Bcell_mock_rep2_R2.fastq.gz
mv SRR2121780_1.fastq.gz WT_Bcell_mock_rep3_R1.fastq.gz
mv SRR2121780_2.fastq.gz WT_Bcell_mock_rep3_R2.fastq.gz
mv SRR2121781_1.fastq.gz WT_Bcell_mock_rep4_R1.fastq.gz
mv SRR2121781_2.fastq.gz WT_Bcell_mock_rep4_R2.fastq.gz
# WT B cells - IR
mv SRR2121786_1.fastq.gz WT_Bcell_IR_rep1_R1.fastq.gz
mv SRR2121786_2.fastq.gz WT_Bcell_IR_rep1_R2.fastq.gz
mv SRR2121787_1.fastq.gz WT_Bcell_IR_rep2_R1.fastq.gz
mv SRR2121787_2.fastq.gz WT_Bcell_IR_rep2_R2.fastq.gz
mv SRR2121788_1.fastq.gz WT_Bcell_IR_rep3_R1.fastq.gz
mv SRR2121788_2.fastq.gz WT_Bcell_IR_rep3_R2.fastq.gz
mv SRR2121789_1.fastq.gz WT_Bcell_IR_rep4_R1.fastq.gz
mv SRR2121789_2.fastq.gz WT_Bcell_IR_rep4_R2.fastq.gzWorkshop data is subsampled
The FASTQ files provided for this workshop have been subsampled to 20 million reads per sample. The original samples contain 100+ million reads each, and running the full pipeline would take many hours—far exceeding our workshop session time.
Never subsample your real experimental data. This is done exclusively for workshop time constraints. Subsampling reduces statistical power and may affect differential expression results.
For reference, here is how the workshop data was prepared. You do not need to run this—the pre-downloaded data is already subsampled.
The original samples from SRA contain 80-100+ million paired-end
reads each. To enable completion of the full analysis pipeline within a
single workshop day, we subsample to 20 million read pairs per sample
using seqtk.
BASH
# Subsample reads to 20 million per sample (WORKSHOP ONLY - do not do this with real data!)
module load biocontainers
module load seqtk
for sample in WT_Bcell_mock_rep1 WT_Bcell_mock_rep2 WT_Bcell_mock_rep3 WT_Bcell_mock_rep4 \
WT_Bcell_IR_rep1 WT_Bcell_IR_rep2 WT_Bcell_IR_rep3 WT_Bcell_IR_rep4; do
# Use same seed for R1 and R2 to maintain pairing
seqtk sample -s 42 ${sample}_R1.fastq.gz 20000000 | gzip > ${sample}_sub_R1.fastq.gz
seqtk sample -s 42 ${sample}_R2.fastq.gz 20000000 | gzip > ${sample}_sub_R2.fastq.gz
done
# Archive original files
mkdir -p original_reads
mv *_R1.fastq.gz *_R2.fastq.gz original_reads/
# Rename subsampled files to original names for pipeline compatibility
for sample in WT_Bcell_mock_rep1 WT_Bcell_mock_rep2 WT_Bcell_mock_rep3 WT_Bcell_mock_rep4 \
WT_Bcell_IR_rep1 WT_Bcell_IR_rep2 WT_Bcell_IR_rep3 WT_Bcell_IR_rep4; do
mv ${sample}_sub_R1.fastq.gz ${sample}_R1.fastq.gz
mv ${sample}_sub_R2.fastq.gz ${sample}_R2.fastq.gz
doneWhy 20 million reads?
- Sufficient depth for differential expression analysis of moderately expressed genes
- Reduces alignment time from ~30 minutes to ~5-10 minutes per sample
- Allows completion of the full workshop pipeline in a single day
- Still produces biologically meaningful results for well-expressed p53 target genes
What you lose by subsampling:
- Statistical power to detect lowly expressed genes
- Precision in abundance estimates
- Ability to detect subtle fold changes
- Some lowly expressed transcripts may appear as zeros
For your own research, always use the full sequencing depth. Modern RNA-seq experiments typically aim for 20-50 million reads for differential expression, but the original unsubsampled data is always preferred.
The directory, after downloading, should look like this:
BASH
data
├── gencode.vM38.primary_assembly.basic.annotation.gtf
├── gencode.vM38.transcripts.fa
├── gencode.vM38.transcripts-clean.fa
├── GRCm39.primary_assembly.genome.fa
├── WT_Bcell_mock_rep1_R1.fastq.gz
├── WT_Bcell_mock_rep1_R2.fastq.gz
├── WT_Bcell_mock_rep2_R1.fastq.gz
├── WT_Bcell_mock_rep2_R2.fastq.gz
├── WT_Bcell_mock_rep3_R1.fastq.gz
├── WT_Bcell_mock_rep3_R2.fastq.gz
├── WT_Bcell_mock_rep4_R1.fastq.gz
├── WT_Bcell_mock_rep4_R2.fastq.gz
├── WT_Bcell_IR_rep1_R1.fastq.gz
├── WT_Bcell_IR_rep1_R2.fastq.gz
├── WT_Bcell_IR_rep2_R1.fastq.gz
├── WT_Bcell_IR_rep2_R2.fastq.gz
├── WT_Bcell_IR_rep3_R1.fastq.gz
├── WT_Bcell_IR_rep3_R2.fastq.gz
├── WT_Bcell_IR_rep4_R1.fastq.gz
├── WT_Bcell_IR_rep4_R2.fastq.gz
└── SRR_Acc_List.txt- RNA-seq requires three main inputs: FASTQ reads, a reference genome (FASTA), and gene annotation (GTF/GFF).
- In lieu of a reference genome and annotation, transcript sequences (FASTA) can be used for transcript-level quantification.
- Keeping files compressed and well-organized supports reproducible analysis.
- Reference files must match in genome build and version.
- Public data repositories such as GEO and SRA provide raw reads and metadata.
- Tools like
wgetandfasterq-dumpenable programmatic, reproducible data retrieval.