Quality control of RNA-seq reads
Last updated on 2026-08-04 | Edit this page
Overview
Questions
- How do we check the quality of raw RNA-seq reads?
- What information do FastQC and MultiQC provide?
- How do we decide if trimming is required?
- What QC issues are common in Illumina RNA-seq data?
Objectives
- Run FastQC and MultiQC on RNA-seq FASTQ files.
- Understand how to interpret the important FastQC modules.
- Learn which FastQC warnings are expected for RNA-seq.
- Understand why trimming is usually unnecessary for standard RNA-seq.
- Identify samples that may require closer inspection.
Introduction
Before mapping reads to a genome, it is important to evaluate the
quality of the raw RNA-seq data.
Quality control helps reveal problems such as adapter contamination, low
quality cycles, and unexpected technical issues.
In this episode, we will run FastQC and MultiQC on the p53/ionizing
radiation dataset and interpret the results.
We will also explain when trimming is useful and when it can be harmful
for alignment based RNA-seq workflows.
FastQC evaluates individual FASTQ files. MultiQC summarizes all FastQC reports into one view, which is helpful for comparing replicates.
Why do we perform QC on FASTQ files?
Quality control answers several questions:
- Are sequencing qualities stable across cycles?
- Are adapters present at high levels?
- Do different replicates show similar patterns?
- Are there outliers that indicate failed libraries?
QC provides confidence that downstream steps will be reliable.
Running FastQC
FastQC generates an HTML report for each FASTQ file. For paired-end datasets, you can run it on both R1 and R2 together or separately.
We will create a directory for QC output and then run FastQC on all FASTQ files.
Run FastQC on your FASTQ files
Use the commands below to run FastQC on all reads in the
data directory.
BASH
cd $SCRATCH/rnaseq-workshop
mkdir -p results/qc_fastq/
sinteractive -A rcac-rnaseq -q standby -p cpu -N 1 -n 4 --time=1:00:00
module load biocontainers
module load fastqc
fastqc data/*.fastq.gz --outdir results/qc_fastq/ --threads 4BASH
results/qc_fastq/
├── WT_Bcell_mock_rep1_R1_fastqc.html
├── WT_Bcell_mock_rep1_R1_fastqc.zip
├── WT_Bcell_mock_rep1_R2_fastqc.html
├── WT_Bcell_mock_rep1_R2_fastqc.zip
├── WT_Bcell_mock_rep2_R1_fastqc.html
├── WT_Bcell_mock_rep2_R1_fastqc.zip
├── WT_Bcell_mock_rep2_R2_fastqc.html
├── WT_Bcell_mock_rep2_R2_fastqc.zip
├── WT_Bcell_mock_rep3_R1_fastqc.html
├── WT_Bcell_mock_rep3_R1_fastqc.zip
├── WT_Bcell_mock_rep3_R2_fastqc.html
├── WT_Bcell_mock_rep3_R2_fastqc.zip
├── WT_Bcell_mock_rep4_R1_fastqc.html
├── WT_Bcell_mock_rep4_R1_fastqc.zip
├── WT_Bcell_mock_rep4_R2_fastqc.html
├── WT_Bcell_mock_rep4_R2_fastqc.zip
├── WT_Bcell_IR_rep1_R1_fastqc.html
├── WT_Bcell_IR_rep1_R1_fastqc.zip
├── WT_Bcell_IR_rep1_R2_fastqc.html
├── WT_Bcell_IR_rep1_R2_fastqc.zip
├── WT_Bcell_IR_rep2_R1_fastqc.html
├── WT_Bcell_IR_rep2_R1_fastqc.zip
├── WT_Bcell_IR_rep2_R2_fastqc.html
├── WT_Bcell_IR_rep2_R2_fastqc.zip
├── WT_Bcell_IR_rep3_R1_fastqc.html
├── WT_Bcell_IR_rep3_R1_fastqc.zip
├── WT_Bcell_IR_rep3_R2_fastqc.html
├── WT_Bcell_IR_rep3_R2_fastqc.zip
├── WT_Bcell_IR_rep4_R1_fastqc.html
├── WT_Bcell_IR_rep4_R1_fastqc.zip
├── WT_Bcell_IR_rep4_R2_fastqc.html
└── WT_Bcell_IR_rep4_R2_fastqc.zipUnderstanding the FastQC report
FastQC produces several diagnostic modules. RNA-seq users often misinterpret certain modules, so we focus on the ones that matter most.
Which FastQC modules are important for RNA-seq?
Most informative modules:
- Per base sequence quality
- Per sequence quality scores
- Per base sequence content
- Per sequence GC content
- Adapter content
- Overrepresented sequences
Modules that commonly show warnings for RNA-seq, even when libraries are fine:
- Per sequence GC content (RNA-seq reflects transcript GC bias, not whole genome)
- K-mer content (poly A tails and priming artifacts often trigger warnings)
Warnings do not always indicate problems. Context and comparison across samples matter more than individual icons.
Key modules explained
Below are guidelines for interpreting the most useful modules.
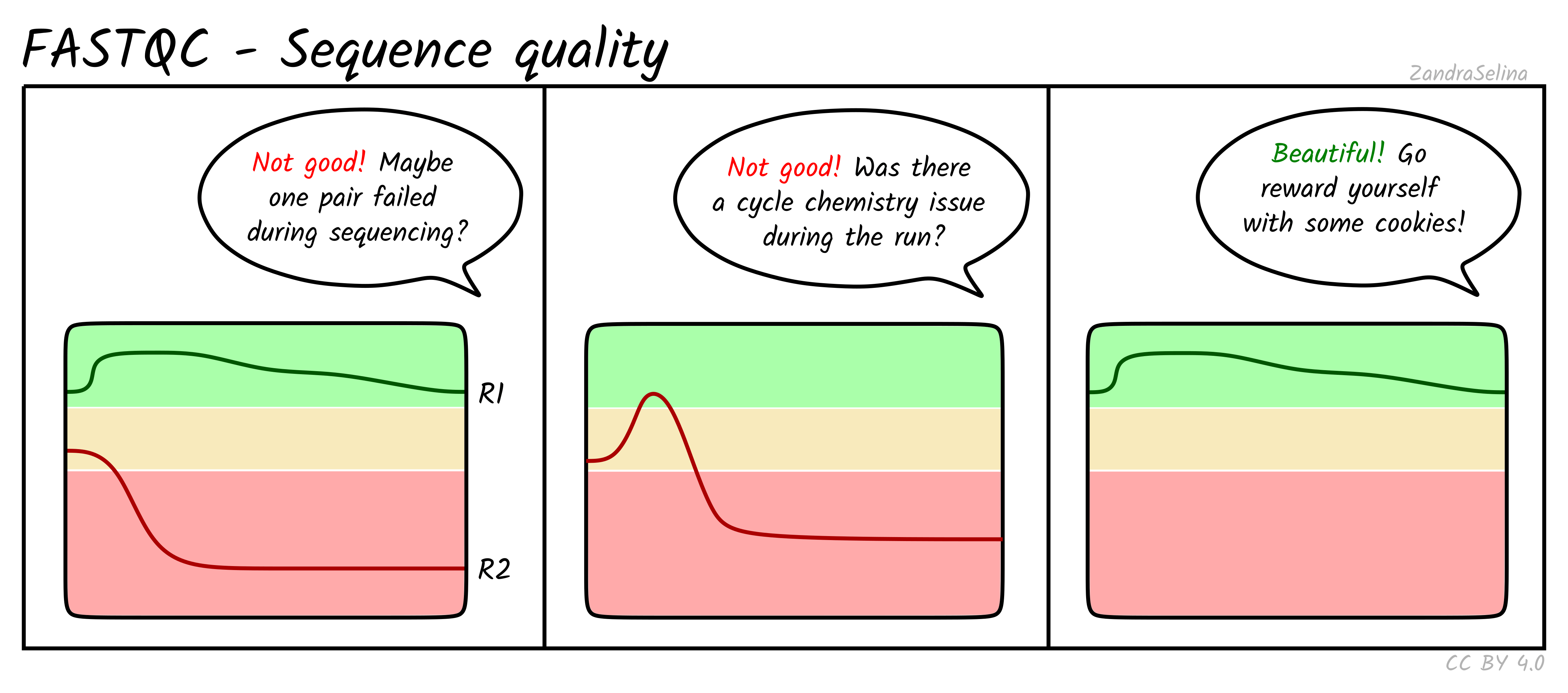
Per base sequence quality
Shows the distribution of quality scores at each position across all reads. You want high and stable quality across the read. A small quality drop at the end of R2 is common and usually not a problem.
Per tile sequence quality
Reports whether specific regions of the flowcell produced lower quality reads. Uneven tiles can indicate instrument issues or bubble formation, but this module rarely shows problems in modern Illumina data.
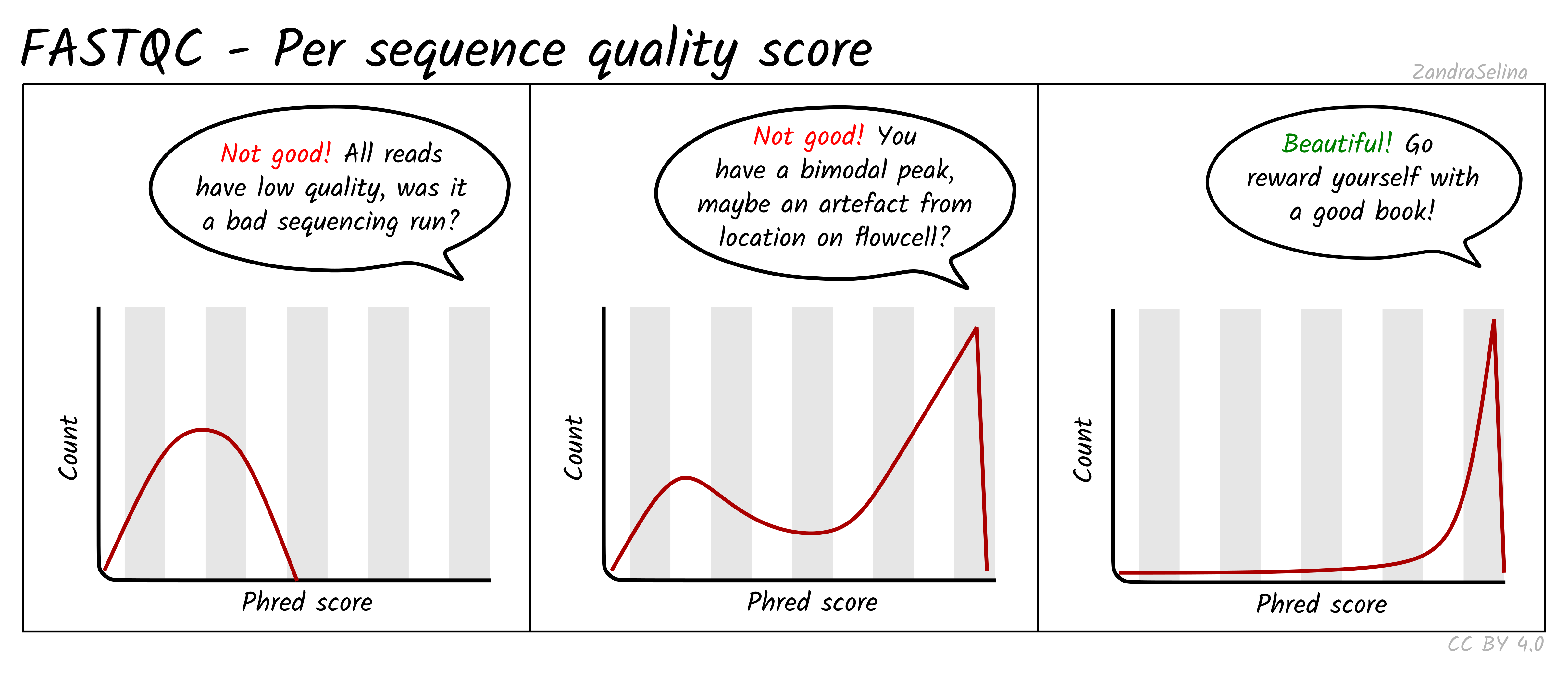
Per sequence quality scores
Shows how many reads have high or low overall quality. A good dataset has most reads with high scores and very few reads with low quality.
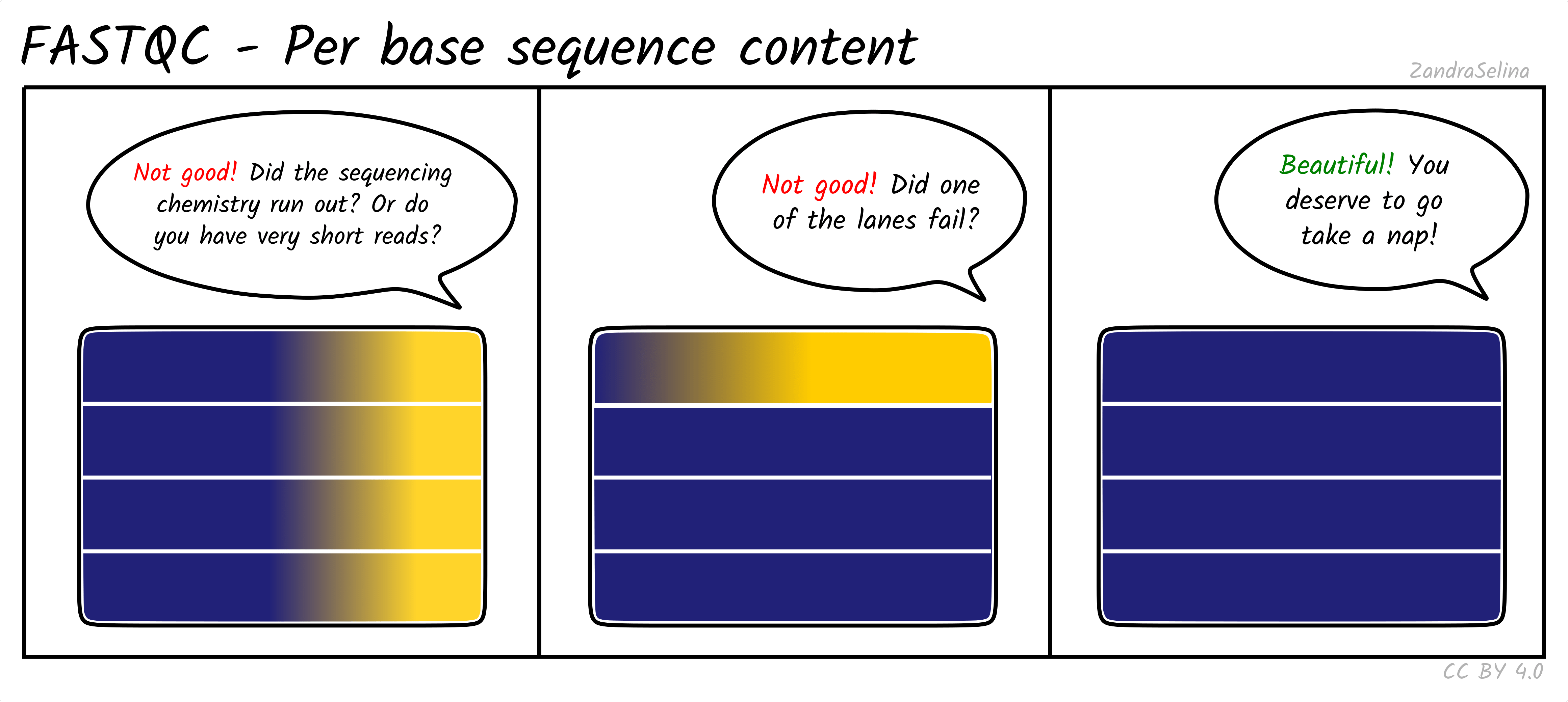
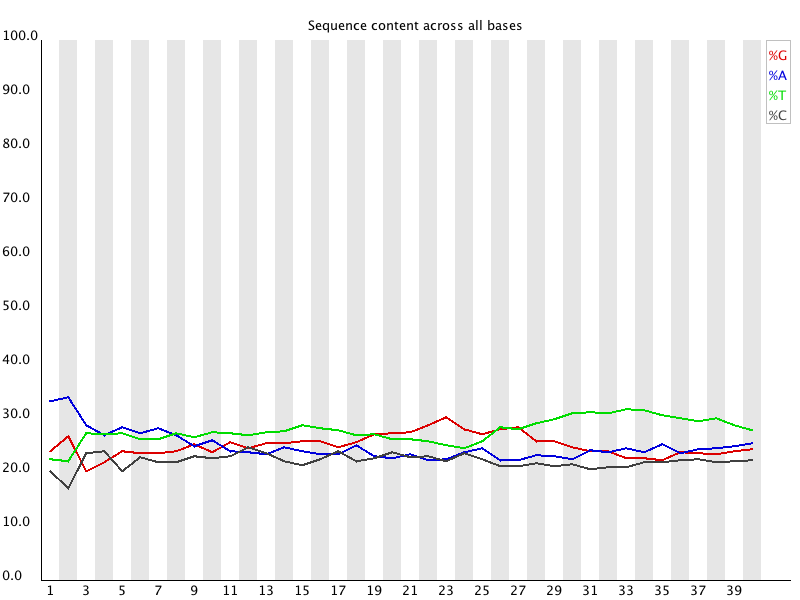
Per base sequence content
Shows the proportion of A, C, G, and T at each cycle. For genomic resequencing, all four lines should be close to 25 percent. RNA-seq usually shows imbalanced composition in early cycles due to biased priming and transcript composition. Mild imbalance at the start of the read is normal and does not require trimming.
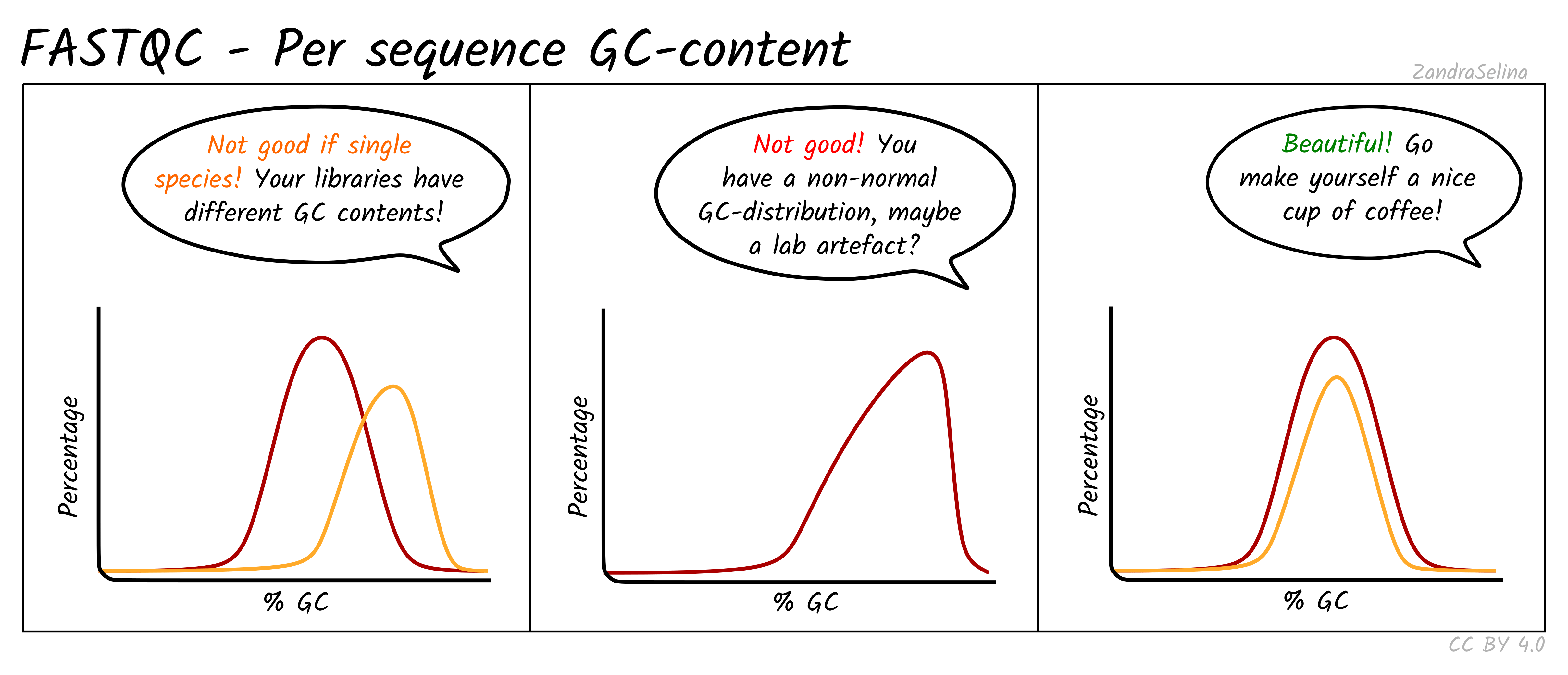
Per sequence GC content
Shows the GC distribution across all reads. RNA-seq often fails this test because expressed transcripts are not GC neutral. Only pronounced multi-modal or extremely shifted distributions are concerning, for example when contamination from another organism is suspected.
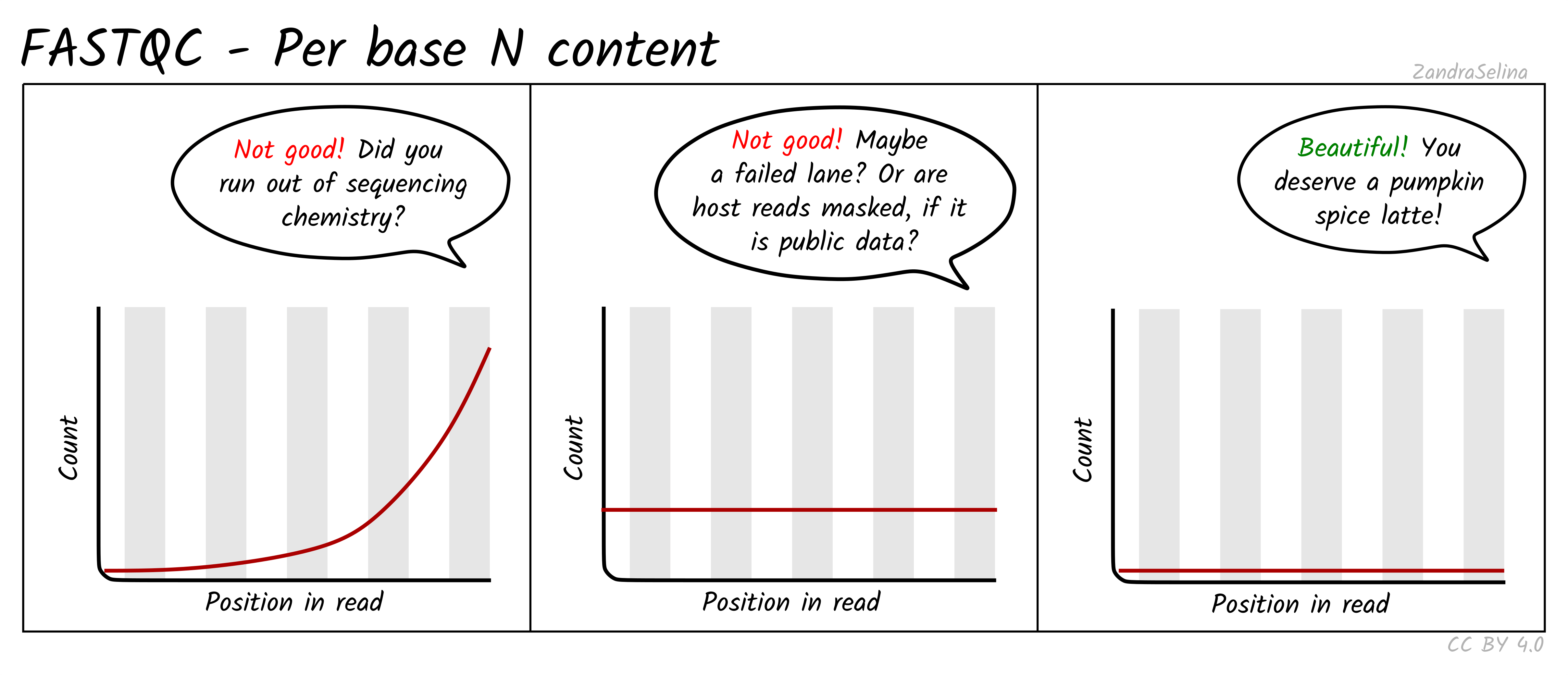
Per base N content
Reports the percentage of unidentified bases (N) at each position. This should be near zero for modern Illumina runs. High N content indicates failed cycles or poor sequencing chemistry.
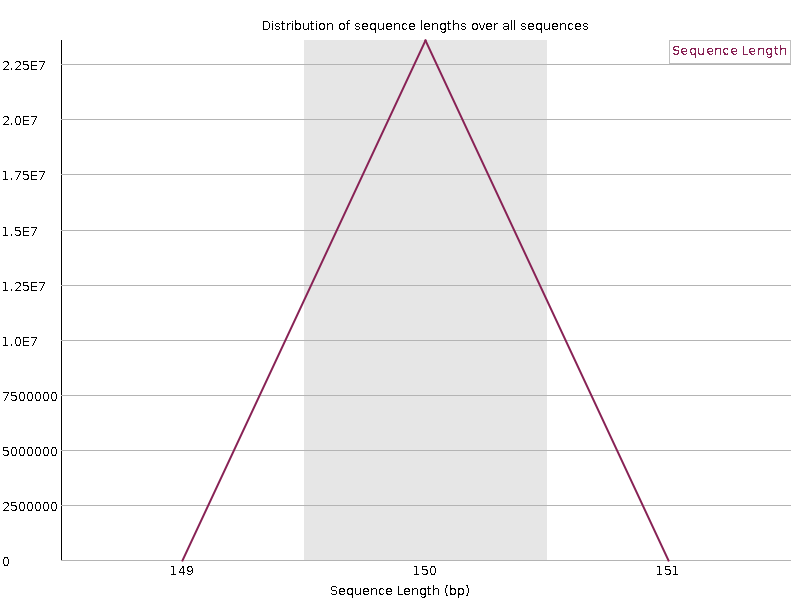
Sequence length distribution
Shows the distribution of read lengths. Standard RNA-seq libraries should have a narrow peak at the expected read length. Variable lengths usually appear after trimming or for specialized protocols such as small RNA.
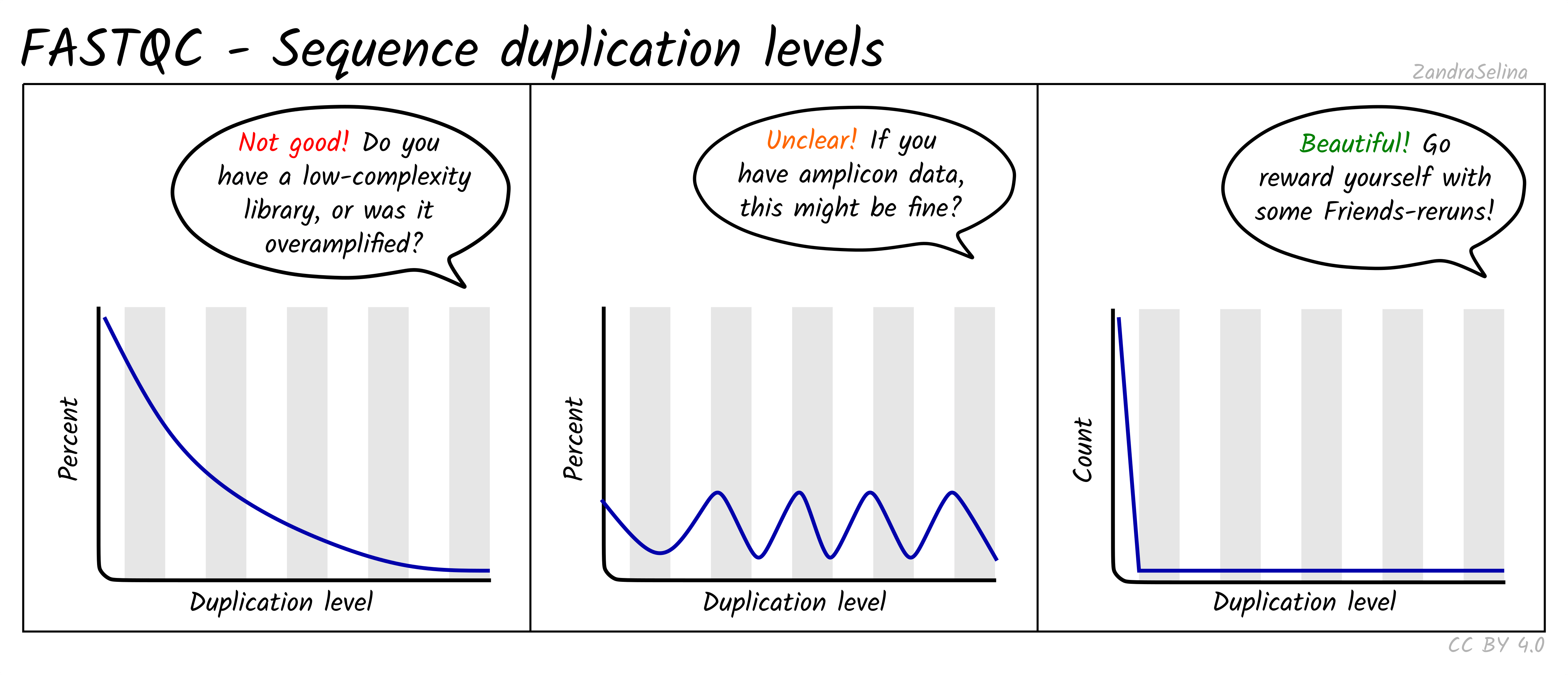
Sequence duplication levels
Reports how many reads are exact duplicates. Some duplication is normal in RNA-seq because highly expressed genes produce many identical fragments. Very high duplication, especially combined with low library size, can indicate low library complexity or overamplification.
Overrepresented sequences
Lists specific sequences that occur more often than expected. Common sources include ribosomal RNA fragments, poly A tails, adapter sequence, or reads from highly expressed transcripts. Some overrepresented sequences are expected, but very high levels may indicate contamination or poor rRNA depletion.
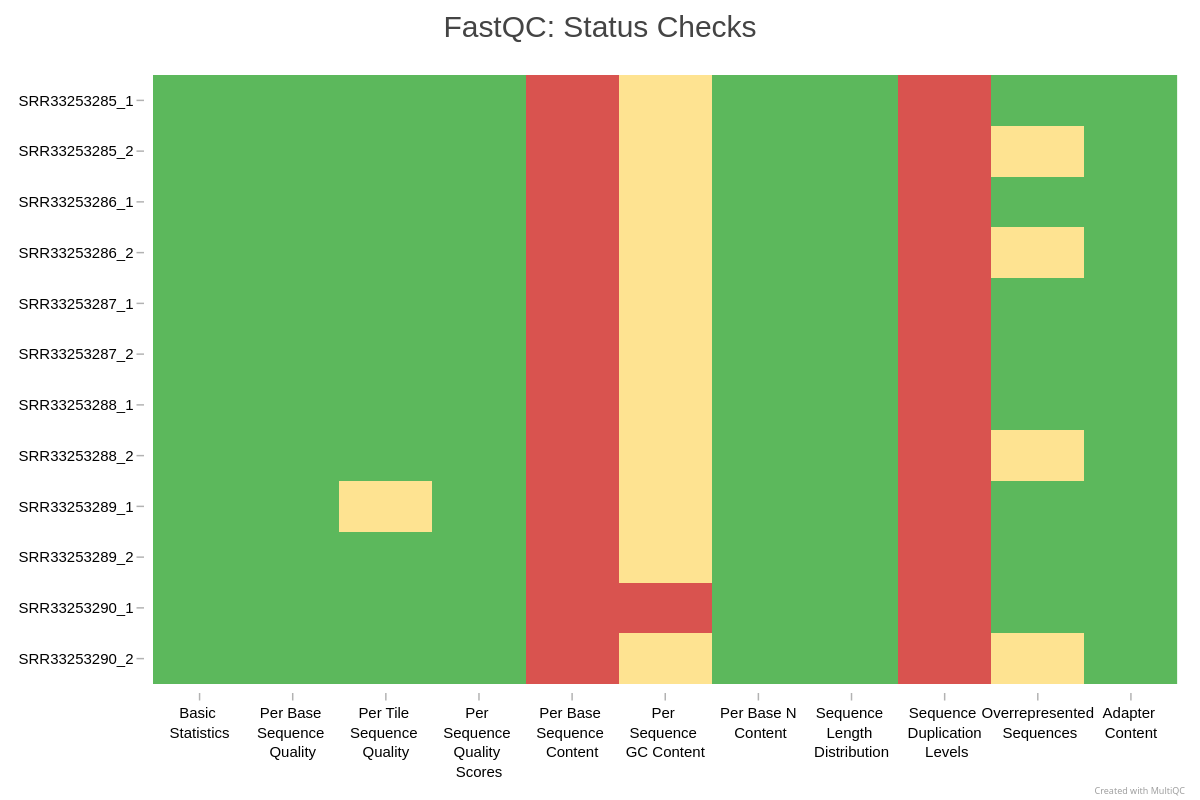
Aggregating reports with MultiQC
FastQC generates a separate HTML file per sample. MultiQC consolidates all reports and allows easy comparison of replicates.
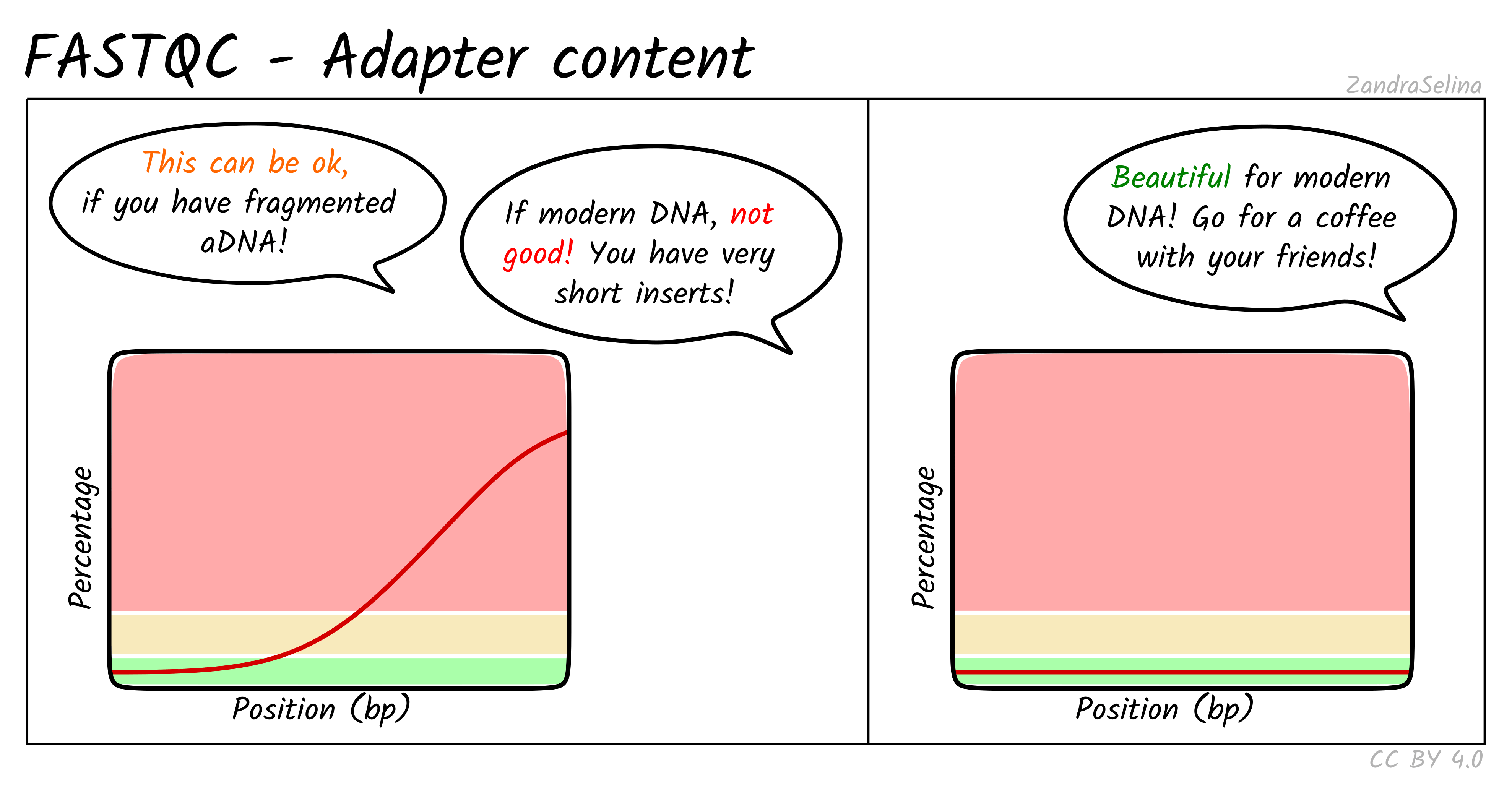
Trimming adapter sequences
Before deciding whether to trim reads, it is essential to understand what adapters are and why they appear in sequencing data.
Adapters are short synthetic DNA sequences ligated to both ends of each fragment during library preparation. They provide primer binding sites needed for cluster amplification and sequencing. Ideally, the sequenced fragment is long enough that the machine reads only biological DNA. However, when the insert is shorter than the read length, the sequencer runs into the adapter sequence, which produces adapter contamination at the ends of reads.
Many users assume that adapters must always be trimmed, but this is not true for typical RNA-seq workflows.
Do we need to trim reads for standard RNA-seq workflows?
For alignment based RNA-seq, adapter trimming is usually not required. Modern aligners such as HISAT2 and STAR detect and soft clip adapter sequence automatically. Soft-clipped bases do not contribute to mismatches or mapping penalties, so adapters generally do not harm alignment quality.
In contrast, unnecessary trimming can introduce new problems:
- shorter reads and lower mapping power
- uneven trimming across samples and artificial differences
- reduction in read complexity
- broken read pairing for paired-end sequencing
- more parameters to track for reproducibility
Trimming is recommended only in specific situations:
- very short inserts, for example degraded RNA or ancient DNA
- adapter sequence dominating read tails, for example more than 10-15 percent adapter content
- transcript assembly workflows such as StringTie or Scallop, which benefit from uniform read lengths
- small RNA or miRNA libraries, where inserts are intentionally very short and adapter presence is guaranteed
Exercise: evaluate the QC results
Inspect your MultiQC report
Using the MultiQC HTML report, answer:
- Do all replicates show similar per base sequence quality profiles?
- Is adapter content high enough to impact analysis?
- Is any sample behaving differently from the rest, for example in number of reads, quality, duplication, or GC percentage?
Typical interpretation for this dataset (students should compare with their own):
- All replicates show high, stable sequence quality.
- Adapter signal appears only at the extreme ends of reads for selected samples and is less than 4 percent.
- No samples show concerning patterns or biases.
- Trimming is therefore unnecessary for this dataset.
Optional reference: how to trim adapters with fastp
(not part of the hands-on)
Sometimes you may encounter datasets where trimming is appropriate, for example short inserts, small RNA, or visibly high adapter content in QC. Although we will not trim reads in this workshop, here is a minimal example you can use in your own projects if trimming becomes necessary.
fastp is a fast, widely used tool that can automatically
detect adapters, trim them, filter low quality reads, and generate QC
reports.
Summary
- FastQC and MultiQC provide essential diagnostics for RNA-seq data.
- Some FastQC warnings are normal for RNA-seq and do not indicate problems.
- Aligners soft clip adapters, so trimming is usually unnecessary for alignment based RNA-seq.
- QC helps detect outliers before alignment and quantification.